Accurate computational design of three-dimensional protein crystals
IF 37.2
1区 材料科学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing arrangements and space group preferences being largely unpredictable. Programming protein crystallization through precisely engineered side-chain–side-chain interactions across protein–protein interfaces is an outstanding challenge. Here we develop a general computational approach for designing three-dimensional protein crystals with prespecified lattice architectures at atomic accuracy that hierarchically constrains the overall number of degrees of freedom of the system. We design three pairs of oligomers that can be individually purified, and upon mixing, spontaneously self-assemble into >100 µm three-dimensional crystals. The structures of these crystals are nearly identical to the computational design models, closely corresponding in both overall architecture and the specific protein–protein interactions. The dimensions of the crystal unit cell can be systematically redesigned while retaining the space group symmetry and overall architecture, and the crystals are extremely porous and highly stable. Our approach enables the computational design of protein crystals with high accuracy, and the designed protein crystals, which have both structural and assembly information encoded in their primary sequences, provide a powerful platform for biological materials engineering. The process of protein crystallization is poorly understood and difficult to program through the primary sequence. Here the authors develop a computational approach to designing three-dimensional protein crystals with prespecified lattice architectures with high accuracy.
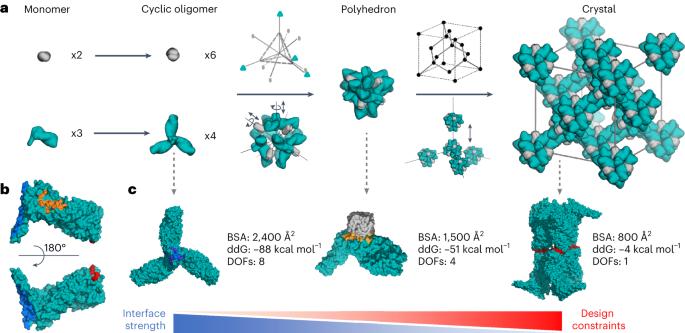
三维蛋白质晶体的精确计算设计。
蛋白质结晶在结构生物学中起着核心作用。尽管如此,结晶过程仍然知之甚少,经验丰富,晶体接触、晶格堆积排列和空间群偏好在很大程度上是不可预测的。通过蛋白质-蛋白质界面上精确设计的侧链-侧链相互作用来编程蛋白质结晶是一个突出的挑战。在这里,我们开发了一种通用的计算方法,用于以原子精度设计具有预先指定的晶格结构的三维蛋白质晶体,该晶格结构在层次上约束系统的总自由度。我们设计了三对低聚物,它们可以单独纯化,混合后自发地自组装成>100 µm三维晶体。这些晶体的结构与计算设计模型几乎相同,在整体结构和特定的蛋白质-蛋白质相互作用方面都非常对应。晶体晶胞的尺寸可以系统地重新设计,同时保持空间群对称性和整体结构,晶体具有极高的多孔性和高度稳定性。我们的方法能够高精度地进行蛋白质晶体的计算设计,所设计的蛋白质晶体在其初级序列中编码了结构和组装信息,为生物材料工程提供了强大的平台。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Materials
工程技术-材料科学:综合
CiteScore
62.20
自引率
0.70%
发文量
221
审稿时长
3.2 months
期刊介绍:
Nature Materials is a monthly multi-disciplinary journal aimed at bringing together cutting-edge research across the entire spectrum of materials science and engineering. It covers all applied and fundamental aspects of the synthesis/processing, structure/composition, properties, and performance of materials. The journal recognizes that materials research has an increasing impact on classical disciplines such as physics, chemistry, and biology.
Additionally, Nature Materials provides a forum for the development of a common identity among materials scientists and encourages interdisciplinary collaboration. It takes an integrated and balanced approach to all areas of materials research, fostering the exchange of ideas between scientists involved in different disciplines.
Nature Materials is an invaluable resource for scientists in academia and industry who are active in discovering and developing materials and materials-related concepts. It offers engaging and informative papers of exceptional significance and quality, with the aim of influencing the development of society in the future.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


