Predictive Modeling and Structure Analysis of Genetic Variants in Familial Hypercholesterolemia: Implications for Diagnosis and Protein Interaction Studies.
Asier Larrea-Sebal, Shifa Jebari-Benslaiman, Unai Galicia-Garcia, Ane San Jose-Urteaga, Kepa B Uribe, Asier Benito-Vicente, César Martín
{"title":"Predictive Modeling and Structure Analysis of Genetic Variants in Familial Hypercholesterolemia: Implications for Diagnosis and Protein Interaction Studies.","authors":"Asier Larrea-Sebal, Shifa Jebari-Benslaiman, Unai Galicia-Garcia, Ane San Jose-Urteaga, Kepa B Uribe, Asier Benito-Vicente, César Martín","doi":"10.1007/s11883-023-01154-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose of review: </strong>Familial hypercholesterolemia (FH) is a hereditary condition characterized by elevated levels of low-density lipoprotein cholesterol (LDL-C), which increases the risk of cardiovascular disease if left untreated. This review aims to discuss the role of bioinformatics tools in evaluating the pathogenicity of missense variants associated with FH. Specifically, it highlights the use of predictive models based on protein sequence, structure, evolutionary conservation, and other relevant features in identifying genetic variants within LDLR, APOB, and PCSK9 genes that contribute to FH.</p><p><strong>Recent findings: </strong>In recent years, various bioinformatics tools have emerged as valuable resources for analyzing missense variants in FH-related genes. Tools such as REVEL, Varity, and CADD use diverse computational approaches to predict the impact of genetic variants on protein function. These tools consider factors such as sequence conservation, structural alterations, and receptor binding to aid in interpreting the pathogenicity of identified missense variants. While these predictive models offer valuable insights, the accuracy of predictions can vary, especially for proteins with unique characteristics that might not be well represented in the databases used for training. This review emphasizes the significance of utilizing bioinformatics tools for assessing the pathogenicity of FH-associated missense variants. Despite their contributions, a definitive diagnosis of a genetic variant necessitates functional validation through in vitro characterization or cascade screening. This step ensures the precise identification of FH-related variants, leading to more accurate diagnoses. Integrating genetic data with reliable bioinformatics predictions and functional validation can enhance our understanding of the genetic basis of FH, enabling improved diagnosis, risk stratification, and personalized treatment for affected individuals. The comprehensive approach outlined in this review promises to advance the management of this inherited disorder, potentially leading to better health outcomes for those affected by FH.</p>","PeriodicalId":10875,"journal":{"name":"Current Atherosclerosis Reports","volume":" ","pages":"839-859"},"PeriodicalIF":5.7000,"publicationDate":"2023-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10618353/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Atherosclerosis Reports","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11883-023-01154-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/17 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PERIPHERAL VASCULAR DISEASE","Score":null,"Total":0}
引用次数: 0
Abstract
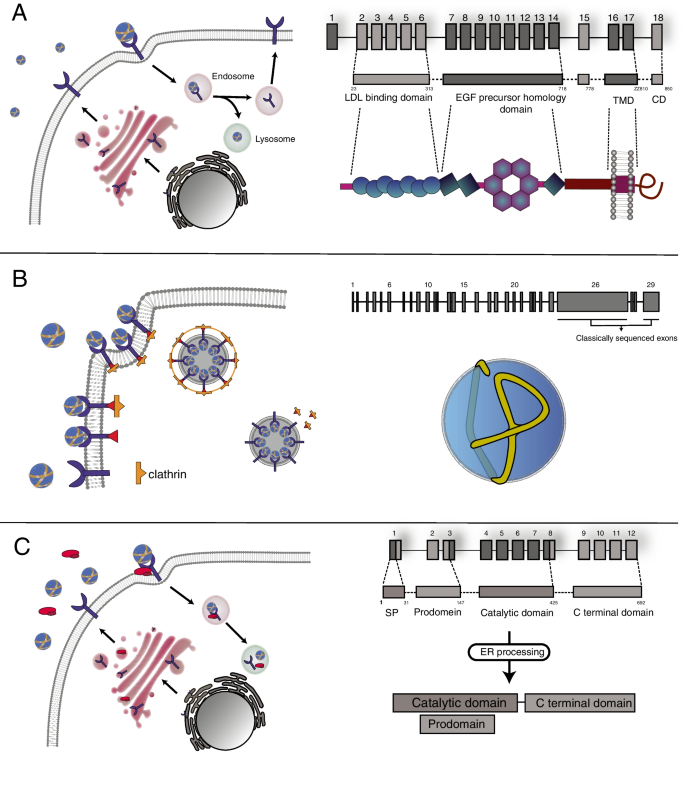
Purpose of review: Familial hypercholesterolemia (FH) is a hereditary condition characterized by elevated levels of low-density lipoprotein cholesterol (LDL-C), which increases the risk of cardiovascular disease if left untreated. This review aims to discuss the role of bioinformatics tools in evaluating the pathogenicity of missense variants associated with FH. Specifically, it highlights the use of predictive models based on protein sequence, structure, evolutionary conservation, and other relevant features in identifying genetic variants within LDLR, APOB, and PCSK9 genes that contribute to FH.
Recent findings: In recent years, various bioinformatics tools have emerged as valuable resources for analyzing missense variants in FH-related genes. Tools such as REVEL, Varity, and CADD use diverse computational approaches to predict the impact of genetic variants on protein function. These tools consider factors such as sequence conservation, structural alterations, and receptor binding to aid in interpreting the pathogenicity of identified missense variants. While these predictive models offer valuable insights, the accuracy of predictions can vary, especially for proteins with unique characteristics that might not be well represented in the databases used for training. This review emphasizes the significance of utilizing bioinformatics tools for assessing the pathogenicity of FH-associated missense variants. Despite their contributions, a definitive diagnosis of a genetic variant necessitates functional validation through in vitro characterization or cascade screening. This step ensures the precise identification of FH-related variants, leading to more accurate diagnoses. Integrating genetic data with reliable bioinformatics predictions and functional validation can enhance our understanding of the genetic basis of FH, enabling improved diagnosis, risk stratification, and personalized treatment for affected individuals. The comprehensive approach outlined in this review promises to advance the management of this inherited disorder, potentially leading to better health outcomes for those affected by FH.
期刊介绍:
The aim of this journal is to systematically provide expert views on current basic science and clinical advances in the field of atherosclerosis and highlight the most important developments likely to transform the field of cardiovascular prevention, diagnosis, and treatment.
We accomplish this aim by appointing major authorities to serve as Section Editors who select leading experts from around the world to provide definitive reviews on key topics and papers published in the past year. We also provide supplementary reviews and commentaries from well-known figures in the field. An Editorial Board of internationally diverse members suggests topics of special interest to their country/region and ensures that topics are current and include emerging research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


