Samrendra K. Singh, Kelsie King, Cole Gannett, Christina Chuong, Soumil Y. Joshi, Charles Plate, Parisa Farzeen, Emily M. Webb, Lakshmi Kumar Kunche, James Weger-Lucarelli, Andrew N. Lowell, Anne M. Brown* and Sanket A. Deshmukh*,
{"title":"Data Driven Computational Design and Experimental Validation of Drugs for Accelerated Mitigation of Pandemic-like Scenarios","authors":"Samrendra K. Singh, Kelsie King, Cole Gannett, Christina Chuong, Soumil Y. Joshi, Charles Plate, Parisa Farzeen, Emily M. Webb, Lakshmi Kumar Kunche, James Weger-Lucarelli, Andrew N. Lowell, Anne M. Brown* and Sanket A. Deshmukh*, ","doi":"10.1021/acs.jpclett.3c01749","DOIUrl":null,"url":null,"abstract":"<p >Emerging pathogens are a historic threat to public health and economic stability. Current trial-and-error approaches to identify new therapeutics are often ineffective due to their inefficient exploration of the enormous small molecule design space. Here, we present a data-driven computational framework composed of hybrid evolutionary algorithms for evolving functional groups on existing drugs to improve their binding affinity toward the main protease (M<sup>pro</sup>) of SARS-CoV-2. We show that combinations of functional groups and sites are critical to design drugs with improved binding affinity, which can be easily achieved using our framework by exploring a fraction of the available search space. Atomistic simulations and experimental validation elucidate that enhanced and prolonged interactions between functionalized drugs and M<sup>pro</sup> residues result in their improved therapeutic value over that of the parental compound. Overall, this novel framework is extremely flexible and has the potential to rapidly design inhibitors for any protein with available crystal structures.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"14 42","pages":"9490–9499"},"PeriodicalIF":4.8000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01749","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
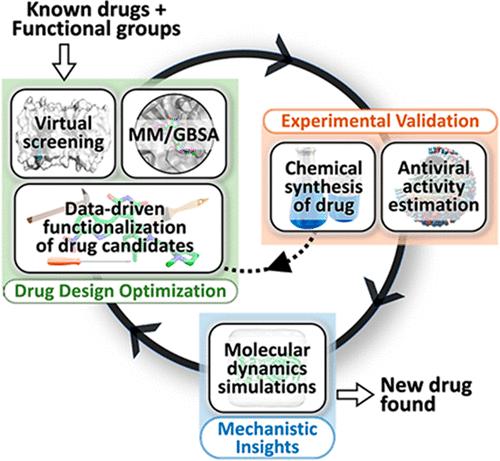
Emerging pathogens are a historic threat to public health and economic stability. Current trial-and-error approaches to identify new therapeutics are often ineffective due to their inefficient exploration of the enormous small molecule design space. Here, we present a data-driven computational framework composed of hybrid evolutionary algorithms for evolving functional groups on existing drugs to improve their binding affinity toward the main protease (Mpro) of SARS-CoV-2. We show that combinations of functional groups and sites are critical to design drugs with improved binding affinity, which can be easily achieved using our framework by exploring a fraction of the available search space. Atomistic simulations and experimental validation elucidate that enhanced and prolonged interactions between functionalized drugs and Mpro residues result in their improved therapeutic value over that of the parental compound. Overall, this novel framework is extremely flexible and has the potential to rapidly design inhibitors for any protein with available crystal structures.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


