Yamin Sun, Min Wang, Wenchao Lin, Wei Dong, Jianguo Xu
{"title":"Massive-scale genomic analysis reveals SARS-CoV-2 mutation characteristics and evolutionary trends.","authors":"Yamin Sun, Min Wang, Wenchao Lin, Wei Dong, Jianguo Xu","doi":"10.1002/mlf2.12040","DOIUrl":null,"url":null,"abstract":"<p><p>The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic resulted in significant societal costs. Hence, an in-depth understanding of SARS-CoV-2 virus mutation and its evolution will help determine the direction of the COVID-19 pandemic. In this study, we identified 296,728 de novo mutations in more than 2,800,000 high-quality SARS-CoV-2 genomes. All possible factors affecting the mutation frequency of SARS-CoV-2 in human hosts were analyzed, including zinc finger antiviral proteins, sequence context, amino acid change, and translation efficiency. As a result, we proposed that when adenine (A) and tyrosine (T) bases are in the context of AM (M stands for adenine or cytosine) or TA motif, A or T base has lower mutation frequency. Furthermore, we hypothesized that translation efficiency can affect the mutation frequency of the third position of the codon by the selection, which explains why SARS-CoV-2 prefers AT3 codons usage. In addition, we found a host-specific asymmetric dinucleotide mutation frequency in the SARS-CoV-2 genome, which provides a new basis for determining the origin of the SARS-CoV-2. Finally, we summarize all possible factors affecting mutation frequency and provide insights into the mutation characteristics and evolutionary trends of SARS-CoV-2.</p>","PeriodicalId":94145,"journal":{"name":"mLife","volume":"1 3","pages":"311-322"},"PeriodicalIF":4.5000,"publicationDate":"2022-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9538474/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"mLife","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/mlf2.12040","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
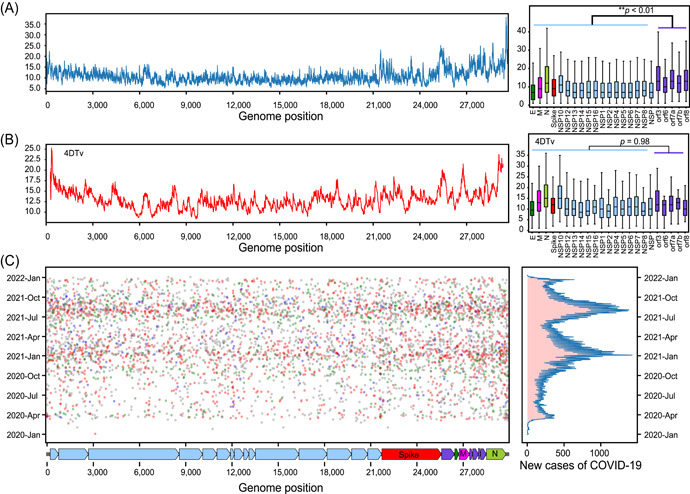
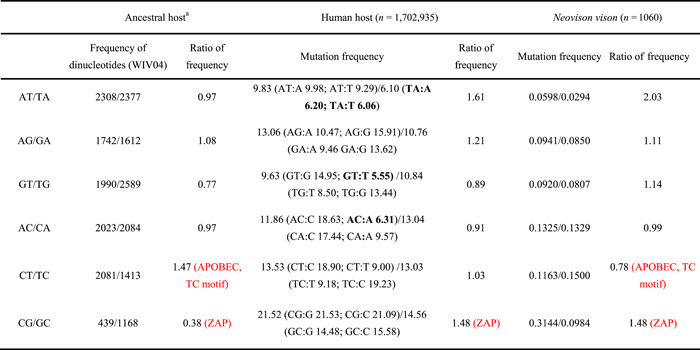
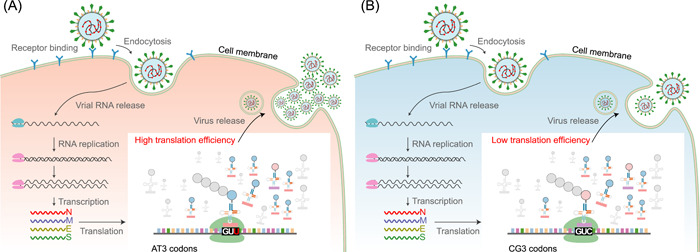
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic resulted in significant societal costs. Hence, an in-depth understanding of SARS-CoV-2 virus mutation and its evolution will help determine the direction of the COVID-19 pandemic. In this study, we identified 296,728 de novo mutations in more than 2,800,000 high-quality SARS-CoV-2 genomes. All possible factors affecting the mutation frequency of SARS-CoV-2 in human hosts were analyzed, including zinc finger antiviral proteins, sequence context, amino acid change, and translation efficiency. As a result, we proposed that when adenine (A) and tyrosine (T) bases are in the context of AM (M stands for adenine or cytosine) or TA motif, A or T base has lower mutation frequency. Furthermore, we hypothesized that translation efficiency can affect the mutation frequency of the third position of the codon by the selection, which explains why SARS-CoV-2 prefers AT3 codons usage. In addition, we found a host-specific asymmetric dinucleotide mutation frequency in the SARS-CoV-2 genome, which provides a new basis for determining the origin of the SARS-CoV-2. Finally, we summarize all possible factors affecting mutation frequency and provide insights into the mutation characteristics and evolutionary trends of SARS-CoV-2.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


