Minha Kim, Krist Aploks, Susana Vargas-Pinto, Xiang Dong
{"title":"RET T244I Germline Variant Mutation in a Patient with Pancreatic Paraganglioma and Primary Hyperparathyroidism.","authors":"Minha Kim, Krist Aploks, Susana Vargas-Pinto, Xiang Dong","doi":"10.5812/ijem-121056","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Paragangliomas are rare neuroendocrine tumors that arise from chromaffin cells. Often termed extra-adrenal pheochromocytomas, these tumors vary with regards to their functionality, location, and malignant potential. Mutations in the RET proto-oncogene are associated with multiple endocrine neoplasia syndrome type 2 (MEN-2) and paragangliomas. The phenotypes of the individual mutations are documented to help determine prognosis.</p><p><strong>Case presentation: </strong>We report a case of a 64-year-old man with a history of parathyroid adenoma who developed a pancreatic retroperitoneal paraganglioma. Despite having laboratory evidence of excess circulating catecholamines, the patient's only presenting symptom was hip pain. The patient underwent resection, and histologic findings were consistent with paraganglioma with lymph node metastasis. Genetic testing revealed a variant of uncertain significance within the RET gene [c.731C>T (p.T244I)].</p><p><strong>Conclusions: </strong>Paragangliomas are rare extra-adrenal neuroendocrine tumors that can be associated with germline mutations. Our patient was diagnosed with a pancreatic paraganglioma associated with a RET T244I mutation. Identifying patients with germline mutations is important for documenting phenotypic presentations of RET gene variants of uncertain significance, which will allow physicians to provide proper management and surveillance of paragangliomas and other associated tumors.</p>","PeriodicalId":13969,"journal":{"name":"International Journal of Endocrinology and Metabolism","volume":"20 3","pages":"e121056"},"PeriodicalIF":1.8000,"publicationDate":"2022-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/22/56/ijem-20-3-121056.PMC9661538.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Endocrinology and Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.5812/ijem-121056","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/7/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Paragangliomas are rare neuroendocrine tumors that arise from chromaffin cells. Often termed extra-adrenal pheochromocytomas, these tumors vary with regards to their functionality, location, and malignant potential. Mutations in the RET proto-oncogene are associated with multiple endocrine neoplasia syndrome type 2 (MEN-2) and paragangliomas. The phenotypes of the individual mutations are documented to help determine prognosis.
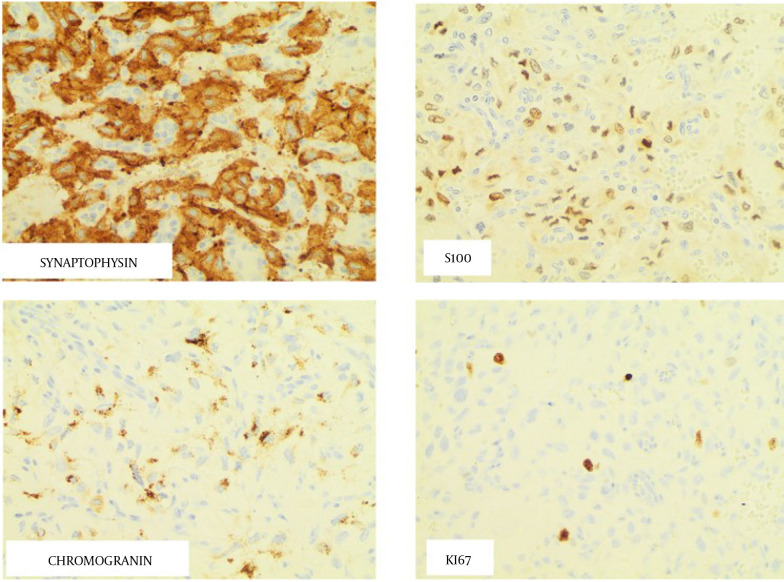
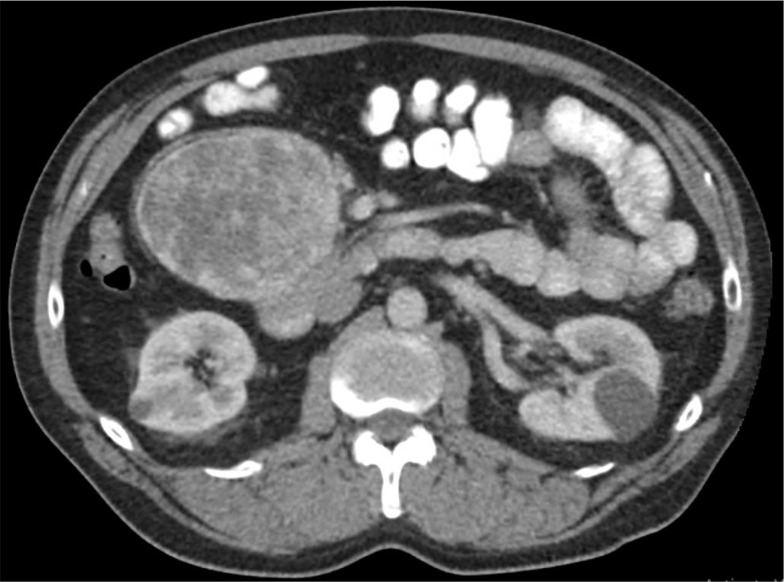
Case presentation: We report a case of a 64-year-old man with a history of parathyroid adenoma who developed a pancreatic retroperitoneal paraganglioma. Despite having laboratory evidence of excess circulating catecholamines, the patient's only presenting symptom was hip pain. The patient underwent resection, and histologic findings were consistent with paraganglioma with lymph node metastasis. Genetic testing revealed a variant of uncertain significance within the RET gene [c.731C>T (p.T244I)].
Conclusions: Paragangliomas are rare extra-adrenal neuroendocrine tumors that can be associated with germline mutations. Our patient was diagnosed with a pancreatic paraganglioma associated with a RET T244I mutation. Identifying patients with germline mutations is important for documenting phenotypic presentations of RET gene variants of uncertain significance, which will allow physicians to provide proper management and surveillance of paragangliomas and other associated tumors.
副神经节瘤是一种罕见的由嗜铬细胞引起的神经内分泌肿瘤。这些肿瘤通常被称为肾上腺外嗜铬细胞瘤,它们在功能、位置和恶性潜能方面各不相同。RET原癌基因突变与多发性内分泌瘤综合征2型(MEN-2)和副神经节瘤有关。记录个体突变的表型以帮助确定预后。病例介绍:我们报告一例64岁男性甲状旁腺瘤病史发展为胰腺腹膜后副神经节瘤。尽管有实验室证据表明循环儿茶酚胺过量,但患者唯一的症状是髋关节疼痛。患者接受了手术切除,组织学结果与伴淋巴结转移的副神经节瘤一致。基因检测显示RET基因中存在一个不确定意义的变异[c]。731 c > T (p.T244I)]。结论:副神经节瘤是一种罕见的肾上腺外神经内分泌肿瘤,可能与种系突变有关。我们的患者被诊断为与RET T244I突变相关的胰腺副神经节瘤。识别生殖系突变患者对于记录不确定意义的RET基因变异的表型表现非常重要,这将使医生能够对副神经节瘤和其他相关肿瘤提供适当的管理和监测。
期刊介绍:
The aim of the International Journal of Endocrinology and Metabolism (IJEM) is to increase knowledge, stimulate research in the field of endocrinology, and promote better management of patients with endocrinological disorders. To achieve this goal, the journal publishes original research papers on human, animal and cell culture studies relevant to endocrinology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


