Novel Homozygous TTI2 Variant Causing Autosomal Recessive Syndromic Intellectual Disability and Primary Microcephaly from Pakistan: A Case Report (Exome Report).
Zul Qarnain, Fatima Khan, Fizza Akbar, Salman Kirmani
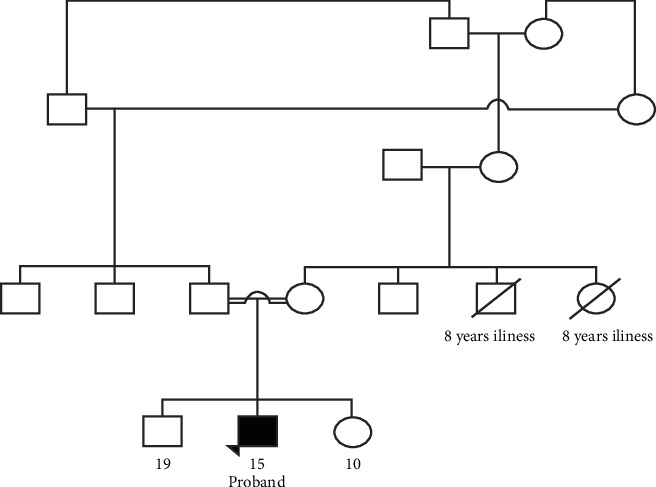
{"title":"Novel Homozygous TTI2 Variant Causing Autosomal Recessive Syndromic Intellectual Disability and Primary Microcephaly from Pakistan: A Case Report (Exome Report).","authors":"Zul Qarnain, Fatima Khan, Fizza Akbar, Salman Kirmani","doi":"10.1155/2022/2766957","DOIUrl":null,"url":null,"abstract":"<p><p>We describe a male patient with a novel TTI2 variant, which has not been previously associated with a human phenotype. His features include intellectual disability, primary microcephaly, delayed psychomotor development, speech delay, short stature, dysmorphic facial features, esotropia, kyphoscoliosis, and behavior abnormalities (Figure). Next generation sequencing revealed autosomal recessive TTI2 variant with uncertain significance, denoted as c.21_22insAAGCGCTCTG (p.Glu8Lysfs × 12). TTI2 encodes a regulator of DNA damage response and helps maintain steady levels of the PIKK family of protein kinases. No disease-causing variants in other genes potentially linked to his clinical presentation were identified. We report a novel loss-of-function homozygous variant in TTI2 that leads to syndromic intellectual disability and primary microcephaly.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":" ","pages":"2766957"},"PeriodicalIF":0.0000,"publicationDate":"2022-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9391182/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2022/2766957","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
We describe a male patient with a novel TTI2 variant, which has not been previously associated with a human phenotype. His features include intellectual disability, primary microcephaly, delayed psychomotor development, speech delay, short stature, dysmorphic facial features, esotropia, kyphoscoliosis, and behavior abnormalities (Figure). Next generation sequencing revealed autosomal recessive TTI2 variant with uncertain significance, denoted as c.21_22insAAGCGCTCTG (p.Glu8Lysfs × 12). TTI2 encodes a regulator of DNA damage response and helps maintain steady levels of the PIKK family of protein kinases. No disease-causing variants in other genes potentially linked to his clinical presentation were identified. We report a novel loss-of-function homozygous variant in TTI2 that leads to syndromic intellectual disability and primary microcephaly.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


