Mahpara Hasan, Gayatra Mainali, Ermal Aliu, Sita Paudel
{"title":"A Case of Autosomal Recessive Intellectual Developmental Disorder Type 5 Presenting with Epilepsy.","authors":"Mahpara Hasan, Gayatra Mainali, Ermal Aliu, Sita Paudel","doi":"10.1155/2022/4056780","DOIUrl":null,"url":null,"abstract":"<p><p>Autosomal recessive intellectual developmental disorder type 5 (MRT5, OMIM # 611091) is caused by biallelic pathogenic variants, leading to loss of function of the NSUN2 gene which encodes a methyltransferase involved in several biological processes, ranging from stress response to neurodevelopment (Hussain 2021). The current literature shows that MRT5 typically manifests with intellectual disability, facial dysmorphism, juvenile cataracts, chronic nephritis, hearing impairment, seizures, cerebellar atrophy, and microcephaly (Pingree et al. 2021). We describe a case of a patient with MRT5 who developed epilepsy in his teens, a rare clinical presentation that has not yet been discussed at length in the literature. Our patient is a 15-year-old male with a history of autism, developmental delay, and focal epilepsy who underwent genetic testing and was found to have a homozygous frameshift mutation in NSUN2 predicted to cause loss of function. This case emphasizes that epilepsy can be a phenotypic manifestation in patients with MRT5.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":" ","pages":"4056780"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9678451/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2022/4056780","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
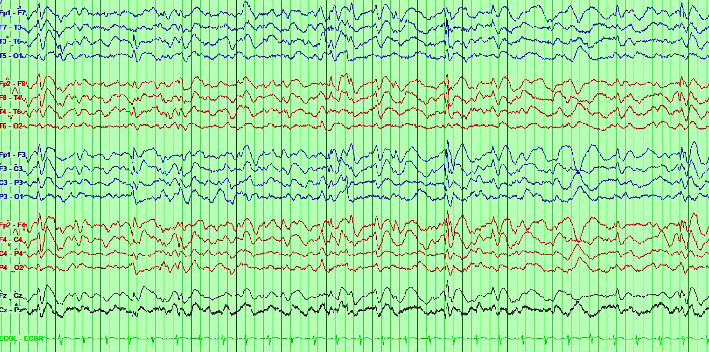
Autosomal recessive intellectual developmental disorder type 5 (MRT5, OMIM # 611091) is caused by biallelic pathogenic variants, leading to loss of function of the NSUN2 gene which encodes a methyltransferase involved in several biological processes, ranging from stress response to neurodevelopment (Hussain 2021). The current literature shows that MRT5 typically manifests with intellectual disability, facial dysmorphism, juvenile cataracts, chronic nephritis, hearing impairment, seizures, cerebellar atrophy, and microcephaly (Pingree et al. 2021). We describe a case of a patient with MRT5 who developed epilepsy in his teens, a rare clinical presentation that has not yet been discussed at length in the literature. Our patient is a 15-year-old male with a history of autism, developmental delay, and focal epilepsy who underwent genetic testing and was found to have a homozygous frameshift mutation in NSUN2 predicted to cause loss of function. This case emphasizes that epilepsy can be a phenotypic manifestation in patients with MRT5.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


