Delayna Willie, Greg Holmes, Ethylin Wang Jabs, Meng Wu
{"title":"Cleft Palate in Apert Syndrome.","authors":"Delayna Willie, Greg Holmes, Ethylin Wang Jabs, Meng Wu","doi":"10.3390/jdb10030033","DOIUrl":null,"url":null,"abstract":"<p><p>Apert syndrome is a rare genetic disorder characterized by craniosynostosis, midface retrusion, and limb anomalies. Cleft palate occurs in a subset of Apert syndrome patients. Although the genetic causes underlying Apert syndrome have been identified, the downstream signaling pathways and cellular mechanisms responsible for cleft palate are still elusive. To find clues for the pathogenic mechanisms of palatal defects in Apert syndrome, we review the clinical characteristics of the palate in cases of Apert syndrome, the palatal phenotypes in mouse models, and the potential signaling mechanisms involved in palatal defects. In Apert syndrome patients, cleft of the soft palate is more frequent than of the hard palate. The length of the hard palate is decreased. Cleft palate is associated most commonly with the S252W variant of FGFR2. In addition to cleft palate, high-arched palate, lateral palatal swelling, or bifid uvula are common in Apert syndrome patients. Mouse models of Apert syndrome display palatal defects, providing valuable tools to understand the underlying mechanisms. The mutations in FGFR2 causing Apert syndrome may change a signaling network in epithelial-mesenchymal interactions during palatogenesis. Understanding the pathogenic mechanisms of palatal defects in Apert syndrome may shed light on potential novel therapeutic solutions.</p>","PeriodicalId":15563,"journal":{"name":"Journal of Developmental Biology","volume":null,"pages":null},"PeriodicalIF":2.2000,"publicationDate":"2022-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9397066/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Developmental Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/jdb10030033","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"DEVELOPMENTAL BIOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
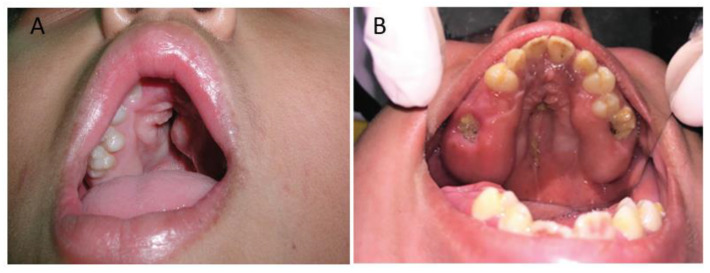
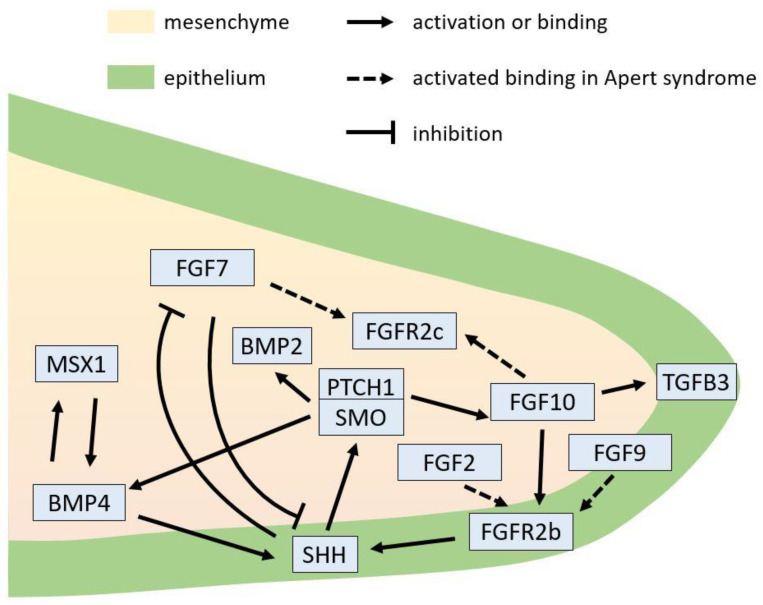
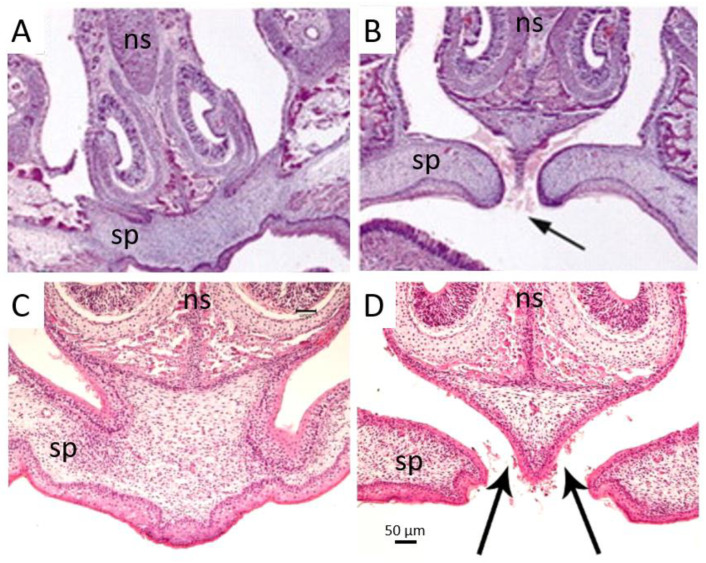
Apert syndrome is a rare genetic disorder characterized by craniosynostosis, midface retrusion, and limb anomalies. Cleft palate occurs in a subset of Apert syndrome patients. Although the genetic causes underlying Apert syndrome have been identified, the downstream signaling pathways and cellular mechanisms responsible for cleft palate are still elusive. To find clues for the pathogenic mechanisms of palatal defects in Apert syndrome, we review the clinical characteristics of the palate in cases of Apert syndrome, the palatal phenotypes in mouse models, and the potential signaling mechanisms involved in palatal defects. In Apert syndrome patients, cleft of the soft palate is more frequent than of the hard palate. The length of the hard palate is decreased. Cleft palate is associated most commonly with the S252W variant of FGFR2. In addition to cleft palate, high-arched palate, lateral palatal swelling, or bifid uvula are common in Apert syndrome patients. Mouse models of Apert syndrome display palatal defects, providing valuable tools to understand the underlying mechanisms. The mutations in FGFR2 causing Apert syndrome may change a signaling network in epithelial-mesenchymal interactions during palatogenesis. Understanding the pathogenic mechanisms of palatal defects in Apert syndrome may shed light on potential novel therapeutic solutions.
期刊介绍:
The Journal of Developmental Biology (ISSN 2221-3759) is an international, peer-reviewed, quick-refereeing, open access journal, which publishes reviews, research papers and communications on the development of multicellular organisms at the molecule, cell, tissue, organ and whole organism levels. Our aim is to encourage researchers to effortlessly publish their new findings or concepts rapidly in an open access medium, overseen by their peers. There is no restriction on the length of the papers; the full experimental details must be provided so that the results can be reproduced. Electronic files regarding the full details of the experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material. Journal of Developmental Biology focuses on: -Development mechanisms and genetics -Cell differentiation -Embryonal development -Tissue/organism growth -Metamorphosis and regeneration of the organisms. It involves many biological fields, such as Molecular biology, Genetics, Physiology, Cell biology, Anatomy, Embryology, Cancer research, Neurobiology, Immunology, Ecology, Evolutionary biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


