{"title":"Prediction of infectivity of SARS-CoV2: Mathematical model with analysis of docking simulation for spike proteins and angiotensin-converting enzyme 2","authors":"Yutaka Takaoka , Aki Sugano , Yoshitomo Morinaga , Mika Ohta , Kenji Miura , Haruyuki Kataguchi , Minoru Kumaoka , Shigemi Kimura , Yoshimasa Maniwa","doi":"10.1016/j.mran.2022.100227","DOIUrl":null,"url":null,"abstract":"<div><h3>Objectives</h3><p>Variants of a coronavirus (SARS-CoV-2) have been spreading in a global pandemic. Improved understanding of the infectivity of future new variants is important so that effective countermeasures against them can be quickly undertaken. In our research reported here, we aimed to predict the infectivity of SARS-CoV-2 by using a mathematical model with molecular simulation analysis, and we used phylogenetic analysis to determine the evolutionary distance of the spike protein gene (S gene) of SARS-CoV-2.</p></div><div><h3>Methods</h3><p>We subjected the six variants and the wild type of spike protein and human angiotensin-converting enzyme 2 (ACE2) to molecular docking simulation analyses to understand the binding affinity of spike protein and ACE2. We then utilized regression analysis of the correlation coefficient of the mathematical model and the infectivity of SARS-CoV-2 to predict infectivity.</p></div><div><h3>Results</h3><p>The evolutionary distance of the S gene correlated with the infectivity of SARS-CoV-2 variants. The calculated biding affinity for the mathematical model obtained with results of molecular docking simulation also correlated with the infectivity of SARS-CoV-2 variants. These results suggest that the data from the docking simulation for the receptor binding domain of variant spike proteins and human ACE2 were valuable for prediction of SARS-CoV-2 infectivity.</p></div><div><h3>Conclusion</h3><p>We developed a mathematical model for prediction of SARS-CoV-2 variant infectivity by using binding affinity obtained via molecular docking and the evolutionary distance of the S gene.</p></div>","PeriodicalId":48593,"journal":{"name":"Microbial Risk Analysis","volume":"22 ","pages":"Article 100227"},"PeriodicalIF":3.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9212987/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Risk Analysis","FirstCategoryId":"93","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2352352222000275","RegionNum":4,"RegionCategory":"环境科学与生态学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENVIRONMENTAL SCIENCES","Score":null,"Total":0}
引用次数: 4
Abstract
Objectives
Variants of a coronavirus (SARS-CoV-2) have been spreading in a global pandemic. Improved understanding of the infectivity of future new variants is important so that effective countermeasures against them can be quickly undertaken. In our research reported here, we aimed to predict the infectivity of SARS-CoV-2 by using a mathematical model with molecular simulation analysis, and we used phylogenetic analysis to determine the evolutionary distance of the spike protein gene (S gene) of SARS-CoV-2.
Methods
We subjected the six variants and the wild type of spike protein and human angiotensin-converting enzyme 2 (ACE2) to molecular docking simulation analyses to understand the binding affinity of spike protein and ACE2. We then utilized regression analysis of the correlation coefficient of the mathematical model and the infectivity of SARS-CoV-2 to predict infectivity.
Results
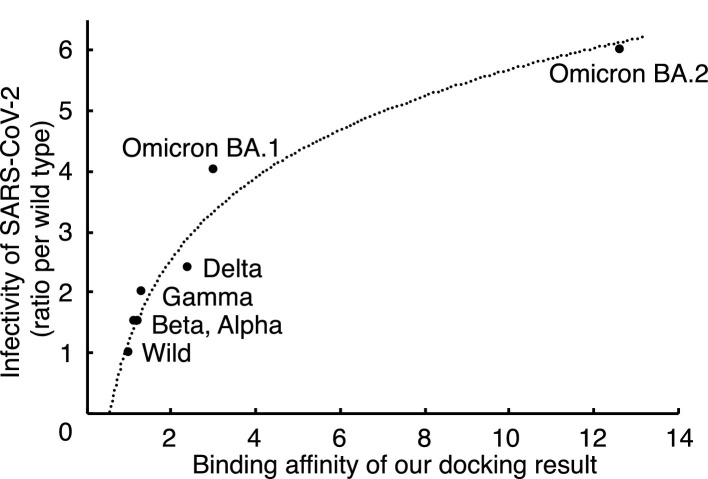
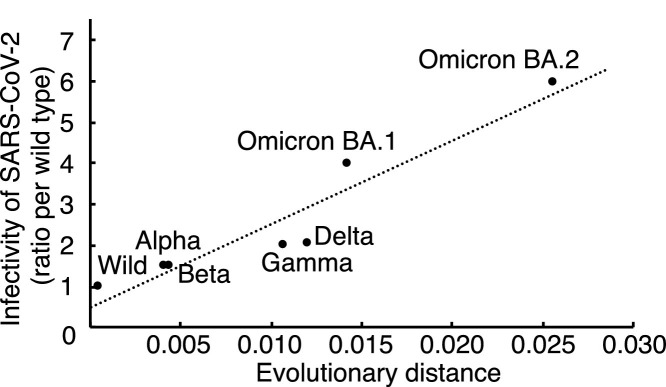
The evolutionary distance of the S gene correlated with the infectivity of SARS-CoV-2 variants. The calculated biding affinity for the mathematical model obtained with results of molecular docking simulation also correlated with the infectivity of SARS-CoV-2 variants. These results suggest that the data from the docking simulation for the receptor binding domain of variant spike proteins and human ACE2 were valuable for prediction of SARS-CoV-2 infectivity.
Conclusion
We developed a mathematical model for prediction of SARS-CoV-2 variant infectivity by using binding affinity obtained via molecular docking and the evolutionary distance of the S gene.
期刊介绍:
The journal Microbial Risk Analysis accepts articles dealing with the study of risk analysis applied to microbial hazards. Manuscripts should at least cover any of the components of risk assessment (risk characterization, exposure assessment, etc.), risk management and/or risk communication in any microbiology field (clinical, environmental, food, veterinary, etc.). This journal also accepts article dealing with predictive microbiology, quantitative microbial ecology, mathematical modeling, risk studies applied to microbial ecology, quantitative microbiology for epidemiological studies, statistical methods applied to microbiology, and laws and regulatory policies aimed at lessening the risk of microbial hazards. Work focusing on risk studies of viruses, parasites, microbial toxins, antimicrobial resistant organisms, genetically modified organisms (GMOs), and recombinant DNA products are also acceptable.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


