Pituitary hypoplasia and growth hormone deficiency in a patient with Coffin-Siris syndrome and severe short stature: case report and literature review.
Stefana Catalina Bilha, Laura Teodoriu, Cristian Velicescu, Lavinia Caba
{"title":"Pituitary hypoplasia and growth hormone deficiency in a patient with Coffin-Siris syndrome and severe short stature: case report and literature review.","authors":"Stefana Catalina Bilha, Laura Teodoriu, Cristian Velicescu, Lavinia Caba","doi":"10.22551/2022.36.0903.10216","DOIUrl":null,"url":null,"abstract":"<p><p>Coffin-Siris syndrome (CSS) is a rare genetic disorder caused by the haploinsufficiency of one of the various genes that are part of the Brahma/BRG1-associated factor (BAF) complex. The BAF complex is one of the chromatin remodeling complexes, involved in embryonic and neural development, and various gene mutations are associated with cognitive impairment. CSS has a highly variable genotype and phenotype expression, thus lacking standardized criteria for diagnosis. It is generally accepted to associate 5<sup>th</sup> digit/nail hypoplasia, intellectual disability (ID)/developmental delay and specific coarse facial features. CSS patients usually display miscellaneous cardiac, genitourinary and central nervous system (CNS) anomalies. Many patients also associate intrauterine growth restriction, failure to thrive and short stature, with several cases demonstrating growth hormone deficiency (GHD). We report the case of a 4-year-old girl with severe short stature (-3.2 standard deviations) due to pituitary hypoplasia and GHD that associated hypoplastic distal phalanx of the 5<sup>th</sup> digit in the hands and feet, severe ID, coarse facial features (bushy eyebrows, bulbous nose, flat nasal bridge, dental anomalies, thick lips, dental anomalies, bilateral epicanthal fold) and CNS anomalies (agenesis of the corpus callosum and bilateral hippocampal atrophy), thus meeting clinical criteria for the diagnosis of CSS. Karyotype was 46,XX. The patient was started on GH replacement therapy, with favorable outcomes. Current practical knowledge regarding CSS diagnosis and management from the endocrinological point of view is also reviewed.</p>","PeriodicalId":72274,"journal":{"name":"Archive of clinical cases","volume":" ","pages":"121-125"},"PeriodicalIF":0.6000,"publicationDate":"2022-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c2/b0/acc-09-03-121.PMC9512126.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archive of clinical cases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22551/2022.36.0903.10216","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 1
Abstract
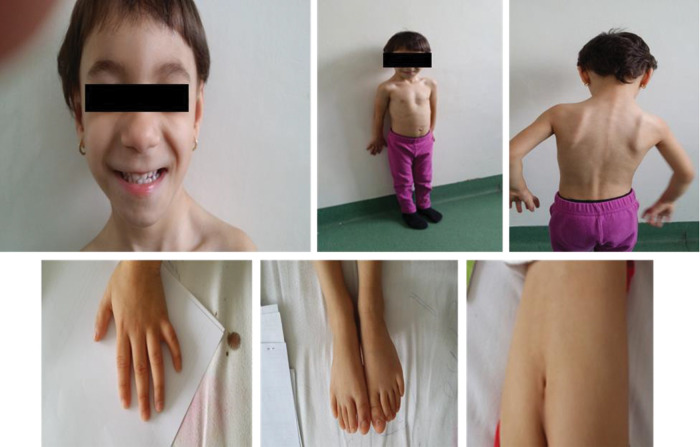
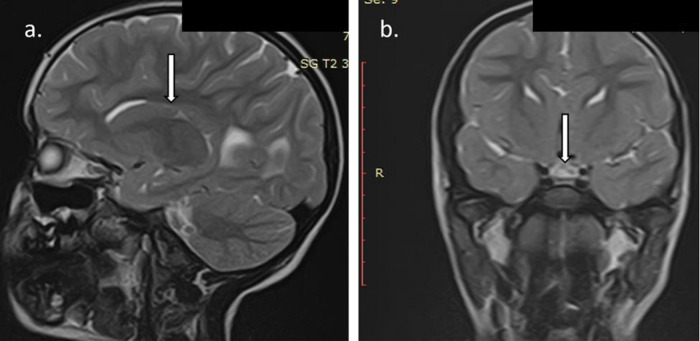
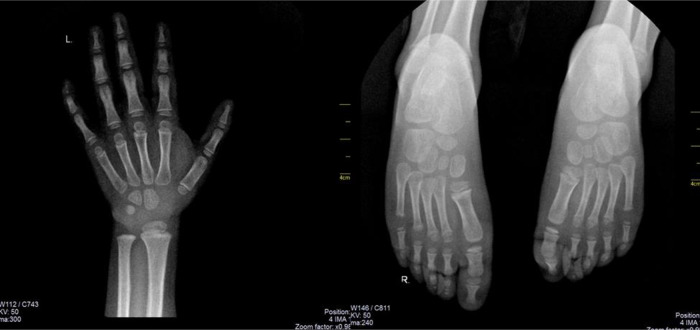
Coffin-Siris syndrome (CSS) is a rare genetic disorder caused by the haploinsufficiency of one of the various genes that are part of the Brahma/BRG1-associated factor (BAF) complex. The BAF complex is one of the chromatin remodeling complexes, involved in embryonic and neural development, and various gene mutations are associated with cognitive impairment. CSS has a highly variable genotype and phenotype expression, thus lacking standardized criteria for diagnosis. It is generally accepted to associate 5th digit/nail hypoplasia, intellectual disability (ID)/developmental delay and specific coarse facial features. CSS patients usually display miscellaneous cardiac, genitourinary and central nervous system (CNS) anomalies. Many patients also associate intrauterine growth restriction, failure to thrive and short stature, with several cases demonstrating growth hormone deficiency (GHD). We report the case of a 4-year-old girl with severe short stature (-3.2 standard deviations) due to pituitary hypoplasia and GHD that associated hypoplastic distal phalanx of the 5th digit in the hands and feet, severe ID, coarse facial features (bushy eyebrows, bulbous nose, flat nasal bridge, dental anomalies, thick lips, dental anomalies, bilateral epicanthal fold) and CNS anomalies (agenesis of the corpus callosum and bilateral hippocampal atrophy), thus meeting clinical criteria for the diagnosis of CSS. Karyotype was 46,XX. The patient was started on GH replacement therapy, with favorable outcomes. Current practical knowledge regarding CSS diagnosis and management from the endocrinological point of view is also reviewed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


