{"title":"Identification of novel mutations among Iranian NPC1 patients: a bioinformatics approach to predict pathogenic mutations.","authors":"Rezvan Abtahi, Parvaneh Karimzadeh, Omid Aryani, Diba Akbarzadeh, Shadab Salehpour, Alireza Rezayi, Seyed Hassan Tonekaboni, Reza Zolfaghari Emameh, Massoud Houshmand","doi":"10.1186/s41065-022-00224-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Niemann-Pick disease type C (NPC) is a rare lysosomal neurovisceral storage disease caused by mutations in the NPC 1 (95%) or NPC2 (5%) genes. The products of NPC1 and NPC2 genes play considerable roles in glycolipid and cholesterol trafficking, which could consequently lead to NPC disease with variable phenotypes displaying a broad spectrum of symptoms.</p><p><strong>Materials: </strong>In the present study 35 Iranian NPC unrelated patients were enrolled. These patients were first analysed by the Filipin Staining test of cholesterol deposits in cells for NPC diagnostics. Genomic DNA was extracted from the samples of peripheral blood leukocytes in EDTA following the manufacturer's protocol. All exon-intron boundaries and coding exons of the NPC1gene were amplified by polymerase chain reaction (PCR) using appropriate sets of primers. Thereafter, the products of PCR were sequenced and analysed using the NCBI database ( https://blast.ncbi.nlm.nih.gov/Blast.cgi ). The variants were reviewed by some databases including the Human Gene Mutation Database (HGMD) ( http://www.hgmd.cf.ac.uk/ac/index.php ) and ClinVar ( https://www.ncbi.nlm.nih.gov/clinvar (. Moreover, all the variants were manually classified in terms of the American College of Medical Genetics and Genomics (ACMG) guideline.</p><p><strong>Results: </strong>The sequence analysis revealed 20 different variations, 10 of which are new, including one nonsense mutation (c.406C > T); three small deletions, (c.3126delC, c.2920_2923delCCTG, and c.2037delG); and six likely pathogenic missense mutations, (c.542C > A, c.1970G > A, c.1993C > G, c.2821 T > C, c.2872C > G, and c.3632 T > A). Finally, the pathogenicity of these new variants was determined using the ACMG guidelines.</p><p><strong>Conclusion: </strong>The present study aimed to facilitate the prenatal diagnosis of NPC patients in the future. In this regard, we identified 10 novel mutations, and verified that the majority of them occurred in six NPC1 exons (5, 8, 9, 13, 19, and 21), that should be considered with a high priority for Iranian patients' cost-effective evaluation.</p>","PeriodicalId":12862,"journal":{"name":"Hereditas","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2022-01-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8793247/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-022-00224-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Niemann-Pick disease type C (NPC) is a rare lysosomal neurovisceral storage disease caused by mutations in the NPC 1 (95%) or NPC2 (5%) genes. The products of NPC1 and NPC2 genes play considerable roles in glycolipid and cholesterol trafficking, which could consequently lead to NPC disease with variable phenotypes displaying a broad spectrum of symptoms.
Materials: In the present study 35 Iranian NPC unrelated patients were enrolled. These patients were first analysed by the Filipin Staining test of cholesterol deposits in cells for NPC diagnostics. Genomic DNA was extracted from the samples of peripheral blood leukocytes in EDTA following the manufacturer's protocol. All exon-intron boundaries and coding exons of the NPC1gene were amplified by polymerase chain reaction (PCR) using appropriate sets of primers. Thereafter, the products of PCR were sequenced and analysed using the NCBI database ( https://blast.ncbi.nlm.nih.gov/Blast.cgi ). The variants were reviewed by some databases including the Human Gene Mutation Database (HGMD) ( http://www.hgmd.cf.ac.uk/ac/index.php ) and ClinVar ( https://www.ncbi.nlm.nih.gov/clinvar (. Moreover, all the variants were manually classified in terms of the American College of Medical Genetics and Genomics (ACMG) guideline.
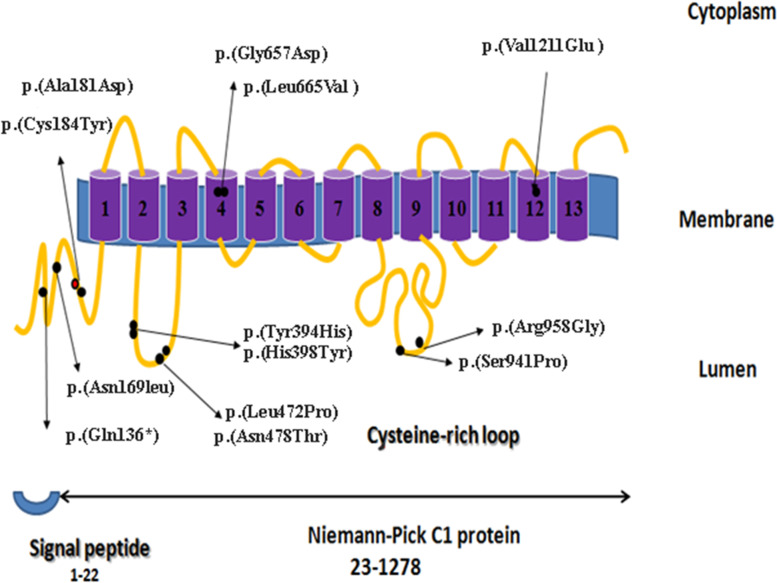
Results: The sequence analysis revealed 20 different variations, 10 of which are new, including one nonsense mutation (c.406C > T); three small deletions, (c.3126delC, c.2920_2923delCCTG, and c.2037delG); and six likely pathogenic missense mutations, (c.542C > A, c.1970G > A, c.1993C > G, c.2821 T > C, c.2872C > G, and c.3632 T > A). Finally, the pathogenicity of these new variants was determined using the ACMG guidelines.
Conclusion: The present study aimed to facilitate the prenatal diagnosis of NPC patients in the future. In this regard, we identified 10 novel mutations, and verified that the majority of them occurred in six NPC1 exons (5, 8, 9, 13, 19, and 21), that should be considered with a high priority for Iranian patients' cost-effective evaluation.
HereditasBiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
3.80
自引率
3.70%
发文量
0
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


