Olga Mucha, Paulina Podkalicka, Katarzyna Kaziród, Emilia Samborowska, Józef Dulak, Agnieszka Łoboda
{"title":"Simvastatin does not alleviate muscle pathology in a mouse model of Duchenne muscular dystrophy.","authors":"Olga Mucha, Paulina Podkalicka, Katarzyna Kaziród, Emilia Samborowska, Józef Dulak, Agnieszka Łoboda","doi":"10.1186/s13395-021-00276-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Duchenne muscular dystrophy (DMD) is an incurable disease, caused by the mutations in the DMD gene, encoding dystrophin, an actin-binding cytoskeletal protein. Lack of functional dystrophin results in muscle weakness, degeneration, and as an outcome cardiac and respiratory failure. As there is still no cure for affected individuals, the pharmacological compounds with the potential to treat or at least attenuate the symptoms of the disease are under constant evaluation. The pleiotropic agents, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, known as statins, have been suggested to exert beneficial effects in the mouse model of DMD. On the other hand, they were also reported to induce skeletal-muscle myopathy. Therefore, we decided to verify the hypothesis that simvastatin may be considered a potential therapeutic agent in DMD.</p><p><strong>Methods: </strong>Several methods including functional assessment of muscle function via grip strength measurement, treadmill test, and single-muscle force estimation, enzymatic assays, histological analysis of muscle damage, gene expression evaluation, and immunofluorescence staining were conducted to study simvastatin-related alterations in the mdx mouse model of DMD.</p><p><strong>Results: </strong>In our study, simvastatin treatment of mdx mice did not result in improved running performance, grip strength, or specific force of the single muscle. Creatine kinase and lactate dehydrogenase activity, markers of muscle injury, were also unaffected by simvastatin delivery in mdx mice. Furthermore, no significant changes in inflammation, fibrosis, and angiogenesis were noted. Despite the decreased percentage of centrally nucleated myofibers in gastrocnemius muscle after simvastatin delivery, no changes were noticed in other regeneration-related parameters. Of note, even an increased rate of necrosis was found in simvastatin-treated mdx mice.</p><p><strong>Conclusion: </strong>In conclusion, our study revealed that simvastatin does not ameliorate DMD pathology.</p>","PeriodicalId":5,"journal":{"name":"ACS Applied Materials & Interfaces","volume":" ","pages":"21"},"PeriodicalIF":8.3000,"publicationDate":"2021-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8414747/pdf/","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Materials & Interfaces","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-021-00276-3","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 12
Abstract
Background: Duchenne muscular dystrophy (DMD) is an incurable disease, caused by the mutations in the DMD gene, encoding dystrophin, an actin-binding cytoskeletal protein. Lack of functional dystrophin results in muscle weakness, degeneration, and as an outcome cardiac and respiratory failure. As there is still no cure for affected individuals, the pharmacological compounds with the potential to treat or at least attenuate the symptoms of the disease are under constant evaluation. The pleiotropic agents, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, known as statins, have been suggested to exert beneficial effects in the mouse model of DMD. On the other hand, they were also reported to induce skeletal-muscle myopathy. Therefore, we decided to verify the hypothesis that simvastatin may be considered a potential therapeutic agent in DMD.
Methods: Several methods including functional assessment of muscle function via grip strength measurement, treadmill test, and single-muscle force estimation, enzymatic assays, histological analysis of muscle damage, gene expression evaluation, and immunofluorescence staining were conducted to study simvastatin-related alterations in the mdx mouse model of DMD.
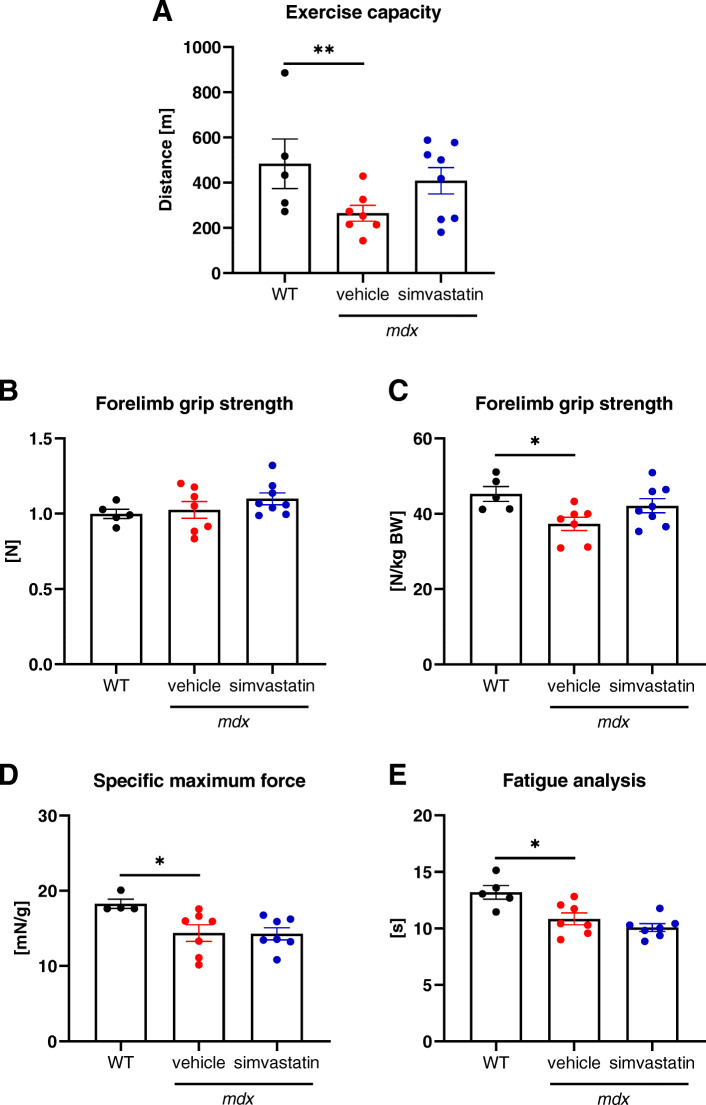
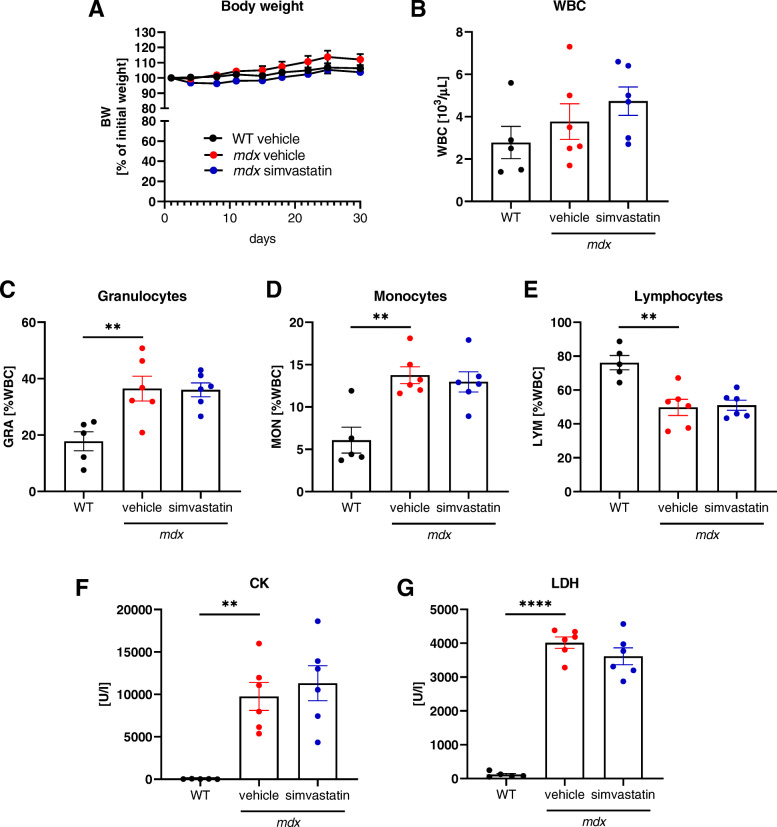
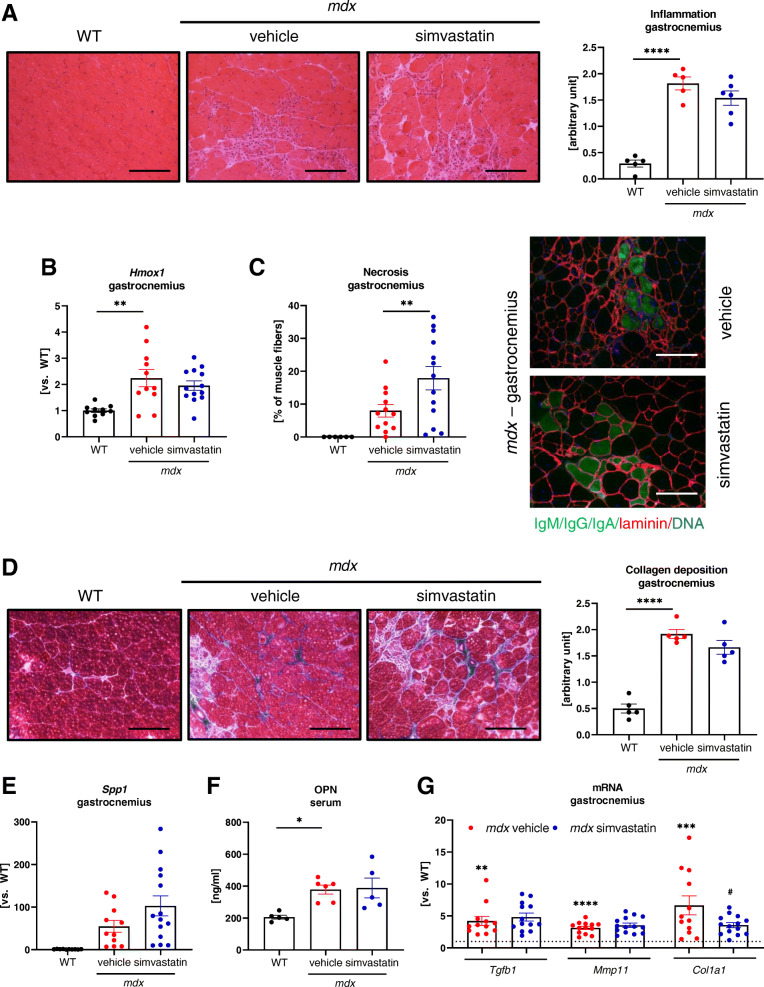
Results: In our study, simvastatin treatment of mdx mice did not result in improved running performance, grip strength, or specific force of the single muscle. Creatine kinase and lactate dehydrogenase activity, markers of muscle injury, were also unaffected by simvastatin delivery in mdx mice. Furthermore, no significant changes in inflammation, fibrosis, and angiogenesis were noted. Despite the decreased percentage of centrally nucleated myofibers in gastrocnemius muscle after simvastatin delivery, no changes were noticed in other regeneration-related parameters. Of note, even an increased rate of necrosis was found in simvastatin-treated mdx mice.
Conclusion: In conclusion, our study revealed that simvastatin does not ameliorate DMD pathology.
期刊介绍:
ACS Applied Materials & Interfaces is a leading interdisciplinary journal that brings together chemists, engineers, physicists, and biologists to explore the development and utilization of newly-discovered materials and interfacial processes for specific applications. Our journal has experienced remarkable growth since its establishment in 2009, both in terms of the number of articles published and the impact of the research showcased. We are proud to foster a truly global community, with the majority of published articles originating from outside the United States, reflecting the rapid growth of applied research worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


