{"title":"Structural relation matching: an algorithm to identify structural patterns into RNAs and their interactions.","authors":"Michela Quadrini","doi":"10.1515/jib-2020-0039","DOIUrl":null,"url":null,"abstract":"<p><p>RNA molecules play crucial roles in various biological processes. Their three-dimensional configurations determine the functions and, in turn, influences the interaction with other molecules. RNAs and their interaction structures, the so-called RNA-RNA interactions, can be abstracted in terms of secondary structures, i.e., a list of the nucleotide bases paired by hydrogen bonding within its nucleotide sequence. Each secondary structure, in turn, can be abstracted into cores and shadows. Both are determined by collapsing nucleotides and arcs properly. We formalize all of these abstractions as arc diagrams, whose arcs determine loops. A secondary structure, represented by an arc diagram, is pseudoknot-free if its arc diagram does not present any crossing among arcs otherwise, it is said pseudoknotted. In this study, we face the problem of identifying a given structural pattern into secondary structures or the associated cores or shadow of both RNAs and RNA-RNA interactions, characterized by arbitrary pseudoknots. These abstractions are mapped into a matrix, whose elements represent the relations among loops. Therefore, we face the problem of taking advantage of matrices and submatrices. The algorithms, implemented in Python, work in polynomial time. We test our approach on a set of 16S ribosomal RNAs with inhibitors of <i>Thermus thermophilus</i>, and we quantify the structural effect of the inhibitors.</p>","PeriodicalId":53625,"journal":{"name":"Journal of Integrative Bioinformatics","volume":"18 2","pages":"111-126"},"PeriodicalIF":1.5000,"publicationDate":"2021-05-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1515/jib-2020-0039","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Integrative Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1515/jib-2020-0039","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
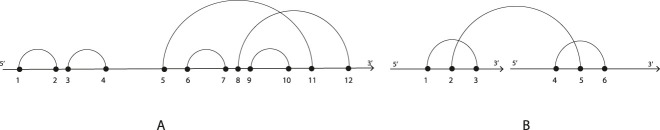
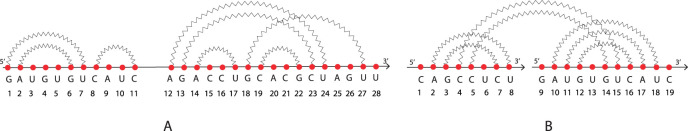
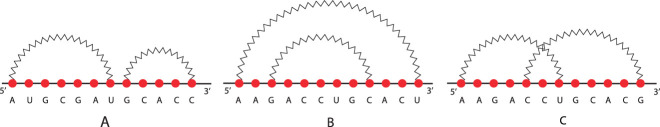
RNA molecules play crucial roles in various biological processes. Their three-dimensional configurations determine the functions and, in turn, influences the interaction with other molecules. RNAs and their interaction structures, the so-called RNA-RNA interactions, can be abstracted in terms of secondary structures, i.e., a list of the nucleotide bases paired by hydrogen bonding within its nucleotide sequence. Each secondary structure, in turn, can be abstracted into cores and shadows. Both are determined by collapsing nucleotides and arcs properly. We formalize all of these abstractions as arc diagrams, whose arcs determine loops. A secondary structure, represented by an arc diagram, is pseudoknot-free if its arc diagram does not present any crossing among arcs otherwise, it is said pseudoknotted. In this study, we face the problem of identifying a given structural pattern into secondary structures or the associated cores or shadow of both RNAs and RNA-RNA interactions, characterized by arbitrary pseudoknots. These abstractions are mapped into a matrix, whose elements represent the relations among loops. Therefore, we face the problem of taking advantage of matrices and submatrices. The algorithms, implemented in Python, work in polynomial time. We test our approach on a set of 16S ribosomal RNAs with inhibitors of Thermus thermophilus, and we quantify the structural effect of the inhibitors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


