{"title":"Pulmonary Alveolar Microlithiasis: A Unique Case of Familial PAM Complicated by Transplant Rejection.","authors":"Austin Helmink, Samir Atiya, Ernesto Martinez Duarte","doi":"10.1155/2021/6674173","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pulmonary alveolar microlithiasis (PAM) is a rare lung disease characterized by the deposition of calcium phosphate microliths or calcospherites, within the alveolar airspace. Typical imaging findings demonstrate a \"sandstorm\" appearance due to bilateral, interstitial sand-like micronodularities with basal predominance.</p><p><strong>Methods and results: </strong>We describe an unusual case of a 48-year-old male with severe, familial PAM ultimately treated with a bilateral lung transplant.</p><p><strong>Conclusions: </strong>PAM is a rare lung disease caused by a mutation in the <i>SLC34A2</i> gene, which encodes for a sodium-phosphate cotransporter in type II alveolar cells, leading to accumulation of intra-alveolar phosphate causing microlith formation. PAM has an indolent course but can progress to chronic hypoxic respiratory failure, ultimately requiring lung transplant, the only known effective treatment.</p>","PeriodicalId":45638,"journal":{"name":"Case Reports in Pathology","volume":"2021 ","pages":"6674173"},"PeriodicalIF":0.5000,"publicationDate":"2021-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8041554/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Pathology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2021/6674173","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"PATHOLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
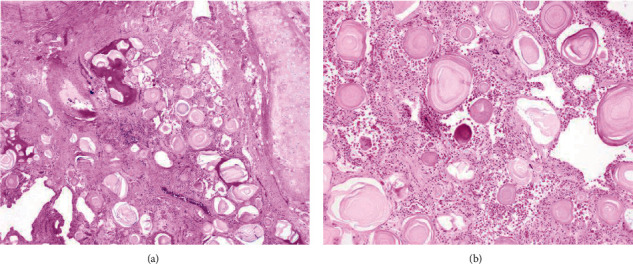
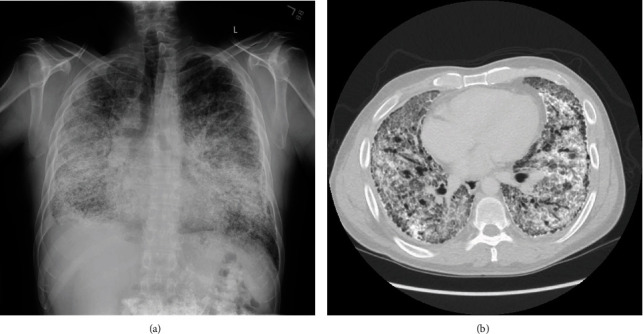
Background: Pulmonary alveolar microlithiasis (PAM) is a rare lung disease characterized by the deposition of calcium phosphate microliths or calcospherites, within the alveolar airspace. Typical imaging findings demonstrate a "sandstorm" appearance due to bilateral, interstitial sand-like micronodularities with basal predominance.
Methods and results: We describe an unusual case of a 48-year-old male with severe, familial PAM ultimately treated with a bilateral lung transplant.
Conclusions: PAM is a rare lung disease caused by a mutation in the SLC34A2 gene, which encodes for a sodium-phosphate cotransporter in type II alveolar cells, leading to accumulation of intra-alveolar phosphate causing microlith formation. PAM has an indolent course but can progress to chronic hypoxic respiratory failure, ultimately requiring lung transplant, the only known effective treatment.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


