Angela L McCall, Justin S Dhindsa, Aidan M Bailey, Logan A Pucci, Laura M Strickland, Mai K ElMallah
{"title":"Glycogen accumulation in smooth muscle of a Pompe disease mouse model.","authors":"Angela L McCall, Justin S Dhindsa, Aidan M Bailey, Logan A Pucci, Laura M Strickland, Mai K ElMallah","doi":"10.1540/jsmr.57.8","DOIUrl":null,"url":null,"abstract":"<p><p>Pompe disease is a lysosomal storage disease caused by mutations within the GAA gene, which encodes acid α-glucosidase (GAA)-an enzyme necessary for lysosomal glycogen degradation. A lack of GAA results in an accumulation of glycogen in cardiac and skeletal muscle, as well as in motor neurons. The only FDA approved treatment for Pompe disease-an enzyme replacement therapy (ERT)-increases survival of patients, but has unmasked previously unrecognized clinical manifestations of Pompe disease. These clinical signs and symptoms include tracheo-bronchomalacia, vascular aneurysms, and gastro-intestinal discomfort. Together, these previously unrecognized pathologies indicate that GAA-deficiency impacts smooth muscle in addition to skeletal and cardiac muscle. Thus, we sought to characterize smooth muscle pathology in the airway, vascular, gastrointestinal, and genitourinary in the Gaa<sup>-/-</sup> mouse model. Increased levels of glycogen were present in smooth muscle cells of the aorta, trachea, esophagus, stomach, and bladder of Gaa<sup>-/-</sup> mice, compared to wild type mice. In addition, there was an increased abundance of both lysosome membrane protein (LAMP1) and autophagosome membrane protein (LC3) indicating vacuolar accumulation in several tissues. Taken together, we show that GAA deficiency results in subsequent pathology in smooth muscle cells, which may lead to life-threatening complications if not properly treated.</p>","PeriodicalId":39619,"journal":{"name":"Journal of Smooth Muscle Research","volume":"57 0","pages":"8-18"},"PeriodicalIF":0.0000,"publicationDate":"2021-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d6/e1/jsmr-57-008.PMC8053439.pdf","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Smooth Muscle Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1540/jsmr.57.8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 4
Abstract
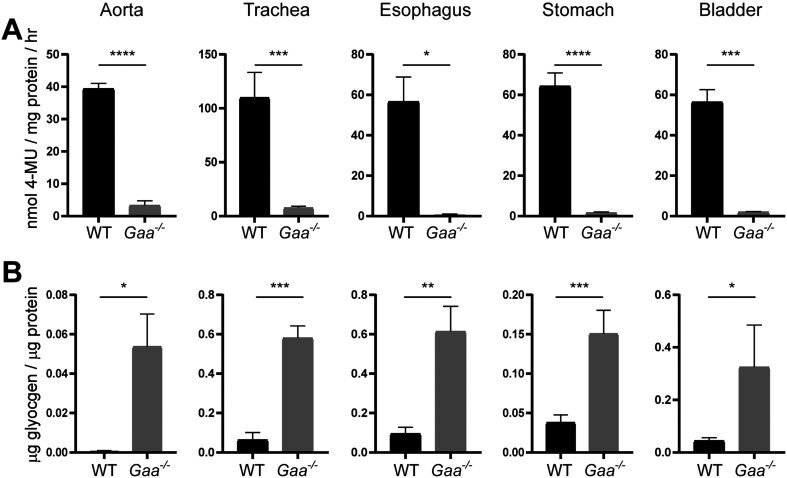
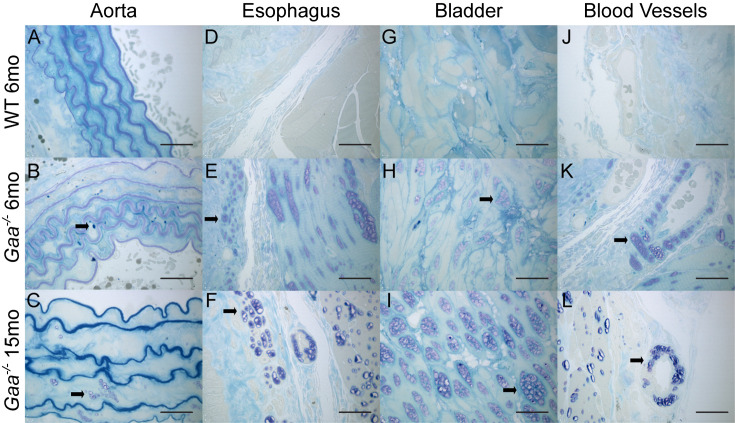
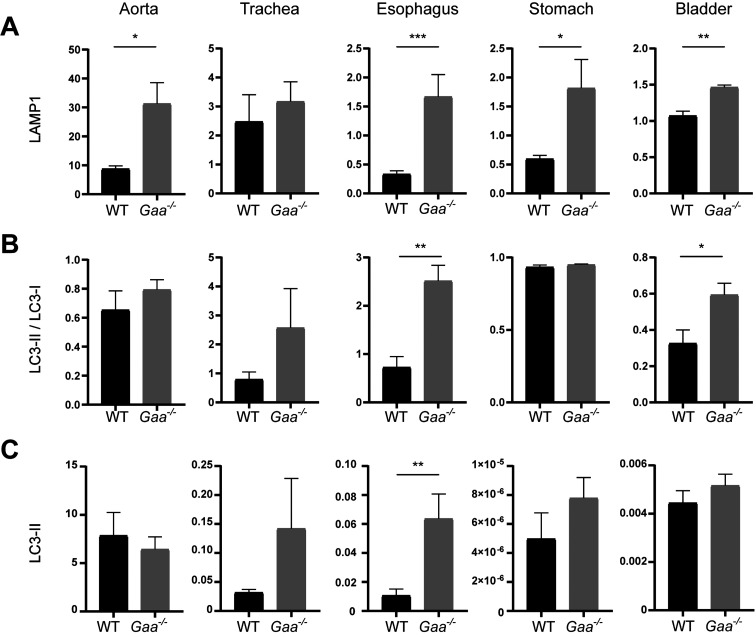
Pompe disease is a lysosomal storage disease caused by mutations within the GAA gene, which encodes acid α-glucosidase (GAA)-an enzyme necessary for lysosomal glycogen degradation. A lack of GAA results in an accumulation of glycogen in cardiac and skeletal muscle, as well as in motor neurons. The only FDA approved treatment for Pompe disease-an enzyme replacement therapy (ERT)-increases survival of patients, but has unmasked previously unrecognized clinical manifestations of Pompe disease. These clinical signs and symptoms include tracheo-bronchomalacia, vascular aneurysms, and gastro-intestinal discomfort. Together, these previously unrecognized pathologies indicate that GAA-deficiency impacts smooth muscle in addition to skeletal and cardiac muscle. Thus, we sought to characterize smooth muscle pathology in the airway, vascular, gastrointestinal, and genitourinary in the Gaa-/- mouse model. Increased levels of glycogen were present in smooth muscle cells of the aorta, trachea, esophagus, stomach, and bladder of Gaa-/- mice, compared to wild type mice. In addition, there was an increased abundance of both lysosome membrane protein (LAMP1) and autophagosome membrane protein (LC3) indicating vacuolar accumulation in several tissues. Taken together, we show that GAA deficiency results in subsequent pathology in smooth muscle cells, which may lead to life-threatening complications if not properly treated.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


