{"title":"An allosteric pocket for inhibition of bacterial Enzyme I identified by NMR-based fragment screening","authors":"Trang T. Nguyen , Vincenzo Venditti","doi":"10.1016/j.yjsbx.2020.100034","DOIUrl":null,"url":null,"abstract":"<div><p>Enzyme I (EI), which is the key enzyme to activate the bacterial phosphotransferase system, plays an important role in the regulation of several metabolic pathways and controls the biology of bacterial cells at multiple levels. The conservation and ubiquity of EI among different types of bacteria makes the enzyme a potential target for antimicrobial research. Here, we use NMR-based fragment screening to identify novel inhibitors of EI. We identify three molecular fragments that allosterically inhibit the phosphoryl transfer reaction catalyzed by EI by interacting with the enzyme at a surface pocket located more than 10 Å away from the substrate binding site. Interestingly, although the three molecules share the same binding pocket, we observe that two of the discovered EI ligands act as competitive inhibitors while the third ligand acts as a mixed inhibitor. Characterization of the EI-inhibitor complexes by NMR and Molecular Dynamics simulations reveals key interactions that perturb the fold of the active site and provides structural foundation for the different inhibitory activity of the identified molecular fragments. In particular, we show that contacts between the inhibitor and the side-chain of V292 are crucial to destabilize binding of the substrate to EI. In contrast, mixed inhibition is caused by additional contacts between the inhibitor and ⍺-helix 2 that perturb the active site structure and turnover in an allosteric manner. We expect our results to provide the basis for the development of second generation allosteric inhibitors of increased potency and to suggest novel molecular strategies to combat drug-resistant infections.</p></div>","PeriodicalId":17238,"journal":{"name":"Journal of Structural Biology: X","volume":"4 ","pages":"Article 100034"},"PeriodicalIF":3.5000,"publicationDate":"2020-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.yjsbx.2020.100034","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Structural Biology: X","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2590152420300167","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 5
Abstract
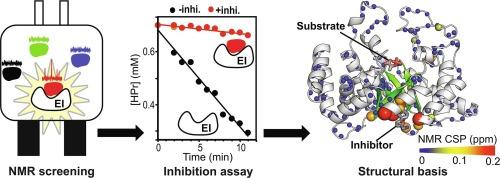
Enzyme I (EI), which is the key enzyme to activate the bacterial phosphotransferase system, plays an important role in the regulation of several metabolic pathways and controls the biology of bacterial cells at multiple levels. The conservation and ubiquity of EI among different types of bacteria makes the enzyme a potential target for antimicrobial research. Here, we use NMR-based fragment screening to identify novel inhibitors of EI. We identify three molecular fragments that allosterically inhibit the phosphoryl transfer reaction catalyzed by EI by interacting with the enzyme at a surface pocket located more than 10 Å away from the substrate binding site. Interestingly, although the three molecules share the same binding pocket, we observe that two of the discovered EI ligands act as competitive inhibitors while the third ligand acts as a mixed inhibitor. Characterization of the EI-inhibitor complexes by NMR and Molecular Dynamics simulations reveals key interactions that perturb the fold of the active site and provides structural foundation for the different inhibitory activity of the identified molecular fragments. In particular, we show that contacts between the inhibitor and the side-chain of V292 are crucial to destabilize binding of the substrate to EI. In contrast, mixed inhibition is caused by additional contacts between the inhibitor and ⍺-helix 2 that perturb the active site structure and turnover in an allosteric manner. We expect our results to provide the basis for the development of second generation allosteric inhibitors of increased potency and to suggest novel molecular strategies to combat drug-resistant infections.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


