Gene regulatory networks on transfer entropy (GRNTE): a novel approach to reconstruct gene regulatory interactions applied to a case study for the plant pathogen Phytophthora infestans.
Juan Camilo Castro, Ivan Valdés, Laura Natalia Gonzalez-García, Giovanna Danies, Silvia Cañas, Flavia Vischi Winck, Carlos Eduardo Ñústez, Silvia Restrepo, Diego Mauricio Riaño-Pachón
{"title":"Gene regulatory networks on transfer entropy (GRNTE): a novel approach to reconstruct gene regulatory interactions applied to a case study for the plant pathogen Phytophthora infestans.","authors":"Juan Camilo Castro, Ivan Valdés, Laura Natalia Gonzalez-García, Giovanna Danies, Silvia Cañas, Flavia Vischi Winck, Carlos Eduardo Ñústez, Silvia Restrepo, Diego Mauricio Riaño-Pachón","doi":"10.1186/s12976-019-0103-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The increasing amounts of genomics data have helped in the understanding of the molecular dynamics of complex systems such as plant and animal diseases. However, transcriptional regulation, although playing a central role in the decision-making process of cellular systems, is still poorly understood. In this study, we linked expression data with mathematical models to infer gene regulatory networks (GRN). We present a simple yet effective method to estimate transcription factors' GRNs from transcriptional data.</p><p><strong>Method: </strong>We defined interactions between pairs of genes (edges in the GRN) as the partial mutual information between these genes that takes into account time and possible lags in time from one gene in relation to another. We call this method Gene Regulatory Networks on Transfer Entropy (GRNTE) and it corresponds to Granger causality for Gaussian variables in an autoregressive model. To evaluate the reconstruction accuracy of our method, we generated several sub-networks from the GRN of the eukaryotic yeast model, Saccharomyces cerevisae. Then, we applied this method using experimental data of the plant pathogen Phytophthora infestans. We evaluated the transcriptional expression levels of 48 transcription factors of P. infestans during its interaction with one moderately resistant and one susceptible cultivar of yellow potato (Solanum tuberosum group Phureja), using RT-qPCR. With these data, we reconstructed the regulatory network of P. infestans during its interaction with these hosts.</p><p><strong>Results: </strong>We first evaluated the performance of our method, based on the transfer entropy (GRNTE), on eukaryotic datasets from the GRNs of the yeast S. cerevisae. Results suggest that GRNTE is comparable with the state-of-the-art methods when the parameters for edge detection are properly tuned. In the case of P. infestans, most of the genes considered in this study, showed a significant change in expression from the onset of the interaction (0 h post inoculum - hpi) to the later time-points post inoculation. Hierarchical clustering of the expression data discriminated two distinct periods during the infection: from 12 to 36 hpi and from 48 to 72 hpi for both the moderately resistant and susceptible cultivars. These distinct periods could be associated with two phases of the life cycle of the pathogen when infecting the host plant: the biotrophic and necrotrophic phases.</p><p><strong>Conclusions: </strong>Here we presented an algorithmic solution to the problem of network reconstruction in time series data. This analytical perspective makes use of the dynamic nature of time series data as it relates to intrinsically dynamic processes such as transcription regulation, were multiple elements of the cell (e.g., transcription factors) act simultaneously and change over time. We applied the algorithm to study the regulatory network of P. infestans during its interaction with two hosts which differ in their level of resistance to the pathogen. Although the gene expression analysis did not show differences between the two hosts, the results of the GRN analyses evidenced rewiring of the genes' interactions according to the resistance level of the host. This suggests that different regulatory processes are activated in response to different environmental cues. Applications of our methodology showed that it could reliably predict where to place edges in the transcriptional networks and sub-networks. The experimental approach used here can help provide insights on the biological role of these interactions on complex processes such as pathogenicity. The code used is available at https://github.com/jccastrog/GRNTE under GNU general public license 3.0.</p>","PeriodicalId":51195,"journal":{"name":"Theoretical Biology and Medical Modelling","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2019-04-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12976-019-0103-7","citationCount":"16","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Biology and Medical Modelling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12976-019-0103-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Mathematics","Score":null,"Total":0}
引用次数: 16
Abstract
Background: The increasing amounts of genomics data have helped in the understanding of the molecular dynamics of complex systems such as plant and animal diseases. However, transcriptional regulation, although playing a central role in the decision-making process of cellular systems, is still poorly understood. In this study, we linked expression data with mathematical models to infer gene regulatory networks (GRN). We present a simple yet effective method to estimate transcription factors' GRNs from transcriptional data.
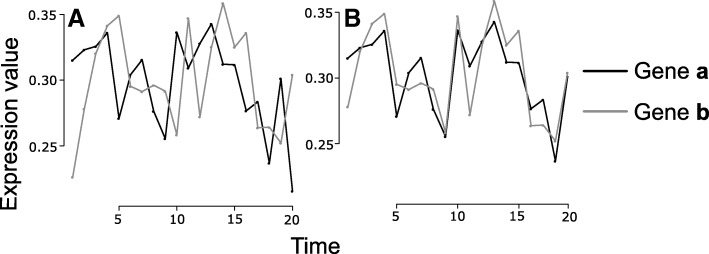
Method: We defined interactions between pairs of genes (edges in the GRN) as the partial mutual information between these genes that takes into account time and possible lags in time from one gene in relation to another. We call this method Gene Regulatory Networks on Transfer Entropy (GRNTE) and it corresponds to Granger causality for Gaussian variables in an autoregressive model. To evaluate the reconstruction accuracy of our method, we generated several sub-networks from the GRN of the eukaryotic yeast model, Saccharomyces cerevisae. Then, we applied this method using experimental data of the plant pathogen Phytophthora infestans. We evaluated the transcriptional expression levels of 48 transcription factors of P. infestans during its interaction with one moderately resistant and one susceptible cultivar of yellow potato (Solanum tuberosum group Phureja), using RT-qPCR. With these data, we reconstructed the regulatory network of P. infestans during its interaction with these hosts.
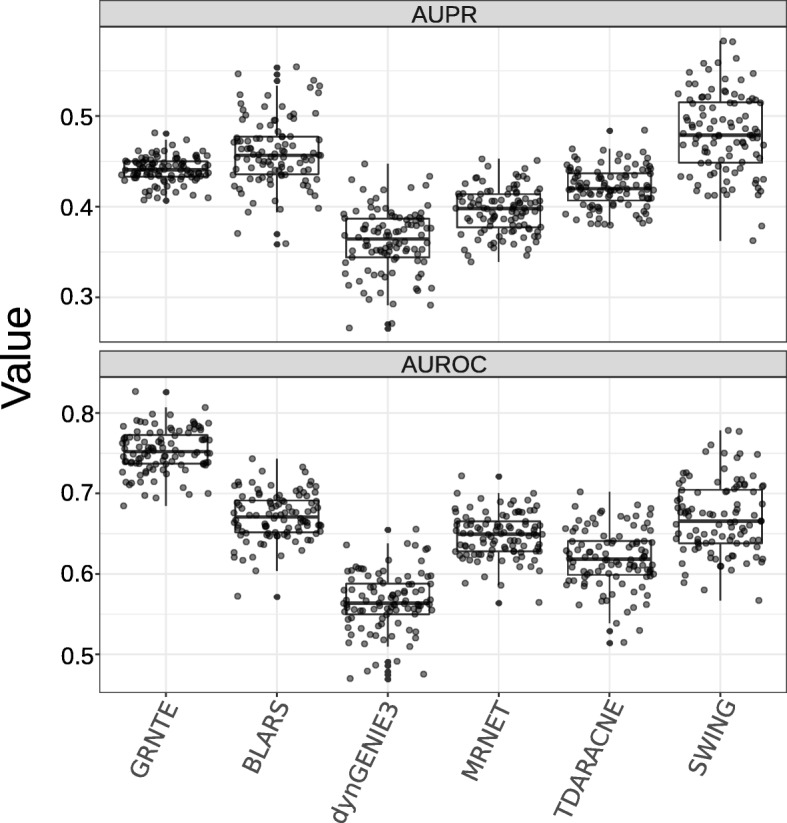
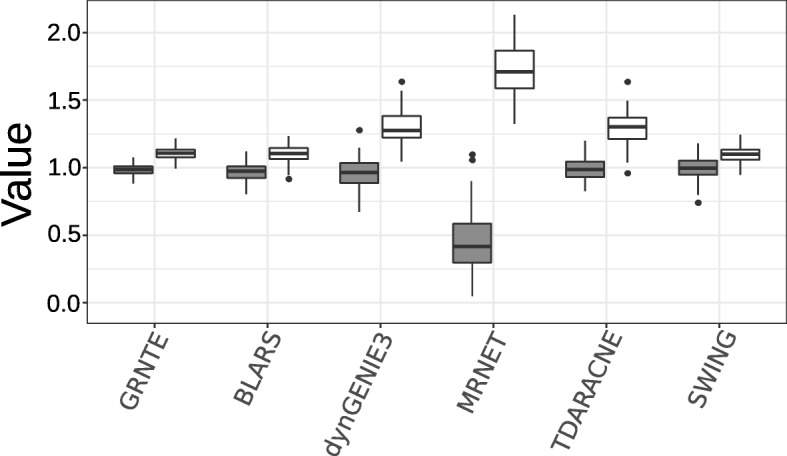
Results: We first evaluated the performance of our method, based on the transfer entropy (GRNTE), on eukaryotic datasets from the GRNs of the yeast S. cerevisae. Results suggest that GRNTE is comparable with the state-of-the-art methods when the parameters for edge detection are properly tuned. In the case of P. infestans, most of the genes considered in this study, showed a significant change in expression from the onset of the interaction (0 h post inoculum - hpi) to the later time-points post inoculation. Hierarchical clustering of the expression data discriminated two distinct periods during the infection: from 12 to 36 hpi and from 48 to 72 hpi for both the moderately resistant and susceptible cultivars. These distinct periods could be associated with two phases of the life cycle of the pathogen when infecting the host plant: the biotrophic and necrotrophic phases.
Conclusions: Here we presented an algorithmic solution to the problem of network reconstruction in time series data. This analytical perspective makes use of the dynamic nature of time series data as it relates to intrinsically dynamic processes such as transcription regulation, were multiple elements of the cell (e.g., transcription factors) act simultaneously and change over time. We applied the algorithm to study the regulatory network of P. infestans during its interaction with two hosts which differ in their level of resistance to the pathogen. Although the gene expression analysis did not show differences between the two hosts, the results of the GRN analyses evidenced rewiring of the genes' interactions according to the resistance level of the host. This suggests that different regulatory processes are activated in response to different environmental cues. Applications of our methodology showed that it could reliably predict where to place edges in the transcriptional networks and sub-networks. The experimental approach used here can help provide insights on the biological role of these interactions on complex processes such as pathogenicity. The code used is available at https://github.com/jccastrog/GRNTE under GNU general public license 3.0.
期刊介绍:
Theoretical Biology and Medical Modelling is an open access peer-reviewed journal adopting a broad definition of "biology" and focusing on theoretical ideas and models associated with developments in biology and medicine. Mathematicians, biologists and clinicians of various specialisms, philosophers and historians of science are all contributing to the emergence of novel concepts in an age of systems biology, bioinformatics and computer modelling. This is the field in which Theoretical Biology and Medical Modelling operates. We welcome submissions that are technically sound and offering either improved understanding in biology and medicine or progress in theory or method.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


