{"title":"Congenital myopathy with hanging big toe due to homozygous myopalladin (MYPN) mutation.","authors":"Luciano Merlini, Patrizia Sabatelli, Manuela Antoniel, Valeria Carinci, Fabio Niro, Giuseppe Monetti, Annalaura Torella, Teresa Giugliano, Cesare Faldini, Vincenzo Nigro","doi":"10.1186/s13395-019-0199-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Myopalladin (MYPN) is a component of the sarcomere that tethers nebulin in skeletal muscle and nebulette in cardiac muscle to alpha-actinin at the Z lines. Autosomal dominant MYPN mutations cause hypertrophic, dilated, or restrictive cardiomyopathy. Autosomal recessive MYPN mutations have been reported in only six families showing a mildly progressive nemaline or cap myopathy with cardiomyopathy in some patients.</p><p><strong>Case presentation: </strong>A consanguineous family with congenital to adult-onset muscle weakness and hanging big toe was reported. Muscle biopsy showed minimal changes with internal nuclei, type 1 fiber predominance, and ultrastructural defects of Z line. Muscle CT imaging showed marked hypodensity of the sartorius bilaterally and MRI scattered abnormal high-intensity areas in the internal tongue muscle and in the posterior cervical muscles. Cardiac involvement was demonstrated by magnetic resonance imaging and late gadolinium enhancement. Whole exome sequencing analysis identified a homozygous loss of function single nucleotide deletion in the exon 11 of the MYPN gene in two siblings. Full-length MYPN protein was undetectable on immunoblotting, and on immunofluorescence, its localization at the Z line was missed.</p><p><strong>Conclusions: </strong>This report extends the phenotypic spectrum of recessive MYPN-related myopathies showing: (1) the two patients had hanging big toe and the oldest one developed spine and hand contractures, none of these signs observed in the previously reported patients, (2) specific ultrastructural changes consisting in Z line fragmentation, but (3) no nemaline or caps on muscle pathology.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":"9 1","pages":"14"},"PeriodicalIF":5.3000,"publicationDate":"2019-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13395-019-0199-9","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-019-0199-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 7
Abstract
Background: Myopalladin (MYPN) is a component of the sarcomere that tethers nebulin in skeletal muscle and nebulette in cardiac muscle to alpha-actinin at the Z lines. Autosomal dominant MYPN mutations cause hypertrophic, dilated, or restrictive cardiomyopathy. Autosomal recessive MYPN mutations have been reported in only six families showing a mildly progressive nemaline or cap myopathy with cardiomyopathy in some patients.
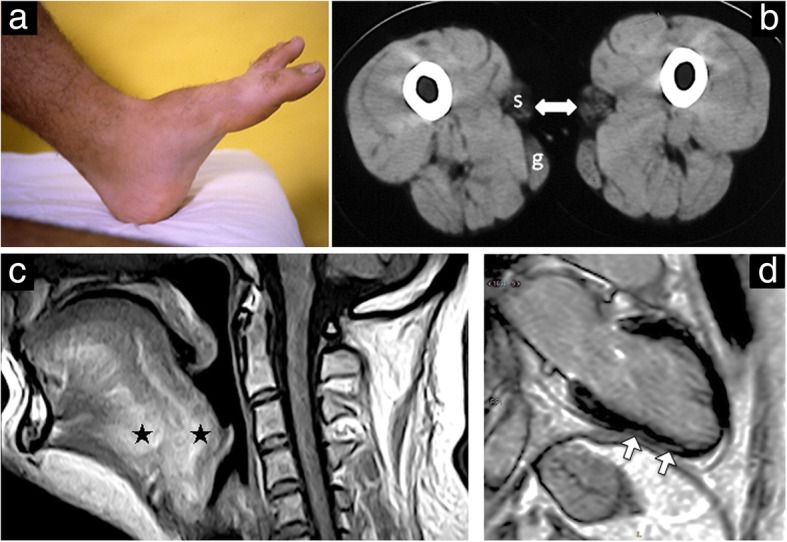
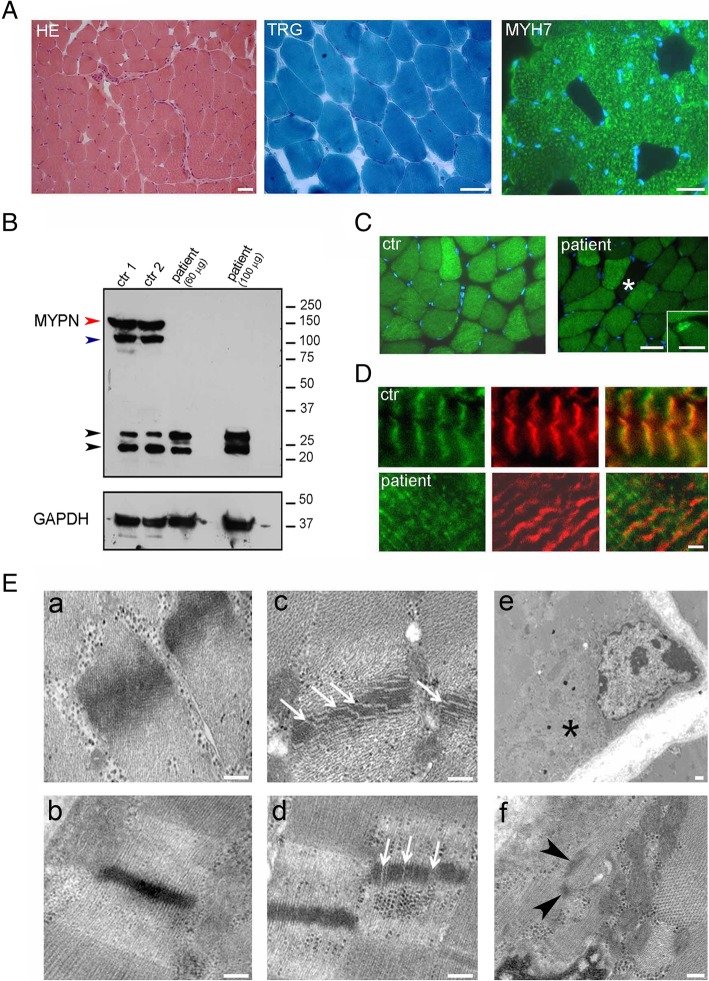
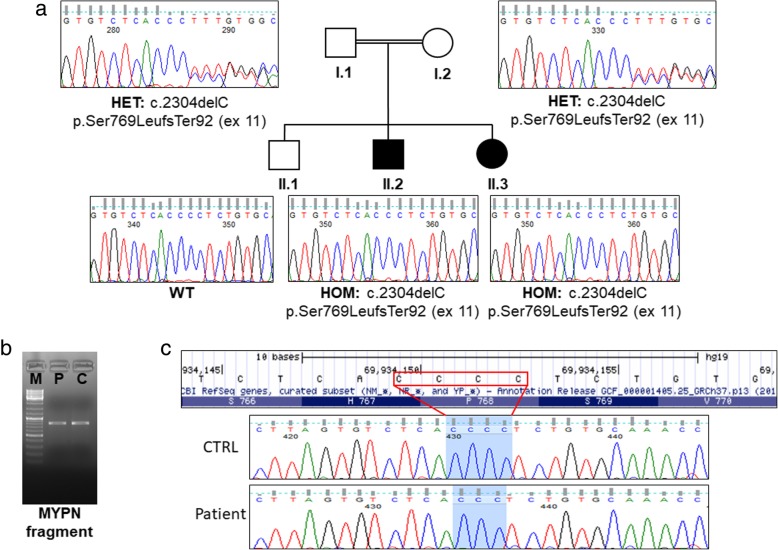
Case presentation: A consanguineous family with congenital to adult-onset muscle weakness and hanging big toe was reported. Muscle biopsy showed minimal changes with internal nuclei, type 1 fiber predominance, and ultrastructural defects of Z line. Muscle CT imaging showed marked hypodensity of the sartorius bilaterally and MRI scattered abnormal high-intensity areas in the internal tongue muscle and in the posterior cervical muscles. Cardiac involvement was demonstrated by magnetic resonance imaging and late gadolinium enhancement. Whole exome sequencing analysis identified a homozygous loss of function single nucleotide deletion in the exon 11 of the MYPN gene in two siblings. Full-length MYPN protein was undetectable on immunoblotting, and on immunofluorescence, its localization at the Z line was missed.
Conclusions: This report extends the phenotypic spectrum of recessive MYPN-related myopathies showing: (1) the two patients had hanging big toe and the oldest one developed spine and hand contractures, none of these signs observed in the previously reported patients, (2) specific ultrastructural changes consisting in Z line fragmentation, but (3) no nemaline or caps on muscle pathology.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


