{"title":"Mitochondrial distribution violation and nuclear indentations in neurons differentiated from iPSCs of Huntington's disease patients.","authors":"Evgeny D Nekrasov, Sergey L Kiselev","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p><b>AIM:</b> Huntington's disease (HD) is an inherited disease caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin gene (<i>HTT</i>) that ultimately leads to neurodegeneration. To study the molecular basis of this disease, induced pluripotent stem cells (iPSCs) generated from patients' fibroblasts were used to investigate axonal mitochondrial trafficking and the nature of nuclear indentations. <b>METHODS:</b> Pathological and control iPSCs generated from patients with a low number of repeats were differentiated in striatal neurons of the brain. Mitochondrial density was measured along the axon using tubulin beta 3 co-staining in pathological and control neurons. To investigate the connection of nuclear roundness with calcium dysregulation, several calcium inhibitors were used. Proteasome system inhibition was applied to mimic premature neuronal ageing. <b>RESULTS:</b> We found that the mitochondrial density was approximately 7.6 ± 0.2 in neurites in control neurons but was only 5.3 ± 0.2 in mutant neurons with 40-44 CAG repeats (p-value <0.005). Neuronal ageing induced by proteasome inhibitor MG132 significantly decreased the mitochondrial density by 15% and 25% in control and mutant neurons to 6.5 ± 0.1 (p-value < 0.005) and 4.0 ± 0.3 (p-value < 0.005), respectively. Thus, for the first time, an impairment of mitochondrial trafficking in pathological neurons with endogenous mutant huntingtin was demonstrated. We found that inhibiting the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the ryanodine-receptor (RyR) or the inositol 1,4,5-trisphosphate receptor (IP3R) by specific inhibitors did not specifically affect the nuclear roundness or survival of pathological neurons differentiated from patient iPSCs. Therefore, nuclear calcium homeostasis is not directly associated with HD pathology. <b>CONCLUSION:</b> Identifying HD iPSCs and differentiating from them neurons provide a unique system for modelling the disease <i>in vitro</i>. Impairments of mitochondrial trafficking and nuclear roundness manifest long before the disease onset, while premature neuronal ageing enhances differences in mitochondrial distribution.</p>","PeriodicalId":17155,"journal":{"name":"Journal of Stem Cells & Regenerative Medicine","volume":"14 2","pages":"80-85"},"PeriodicalIF":1.6000,"publicationDate":"2018-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6339978/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Stem Cells & Regenerative Medicine","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"CELL & TISSUE ENGINEERING","Score":null,"Total":0}
引用次数: 0
Abstract
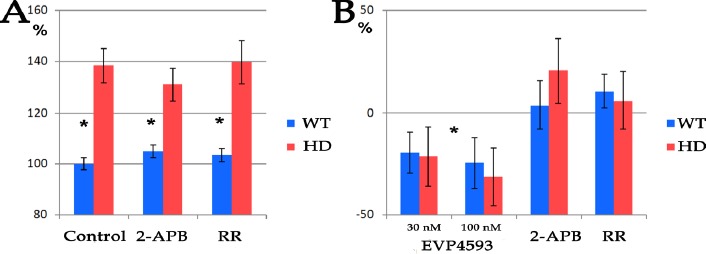
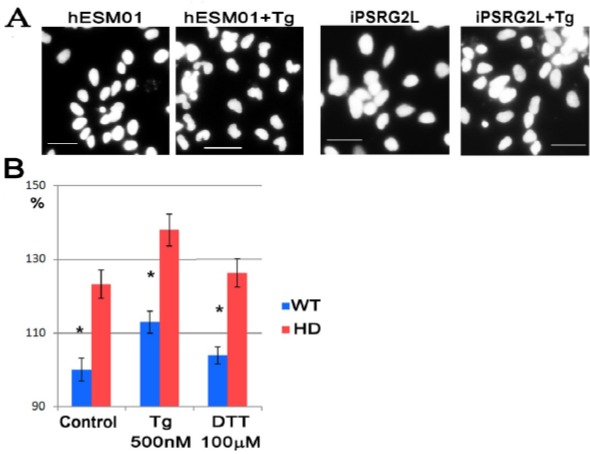
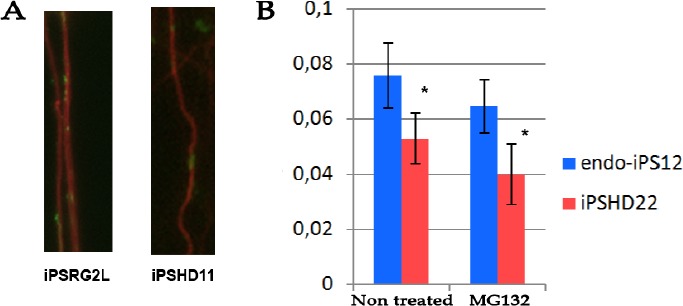
AIM: Huntington's disease (HD) is an inherited disease caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin gene (HTT) that ultimately leads to neurodegeneration. To study the molecular basis of this disease, induced pluripotent stem cells (iPSCs) generated from patients' fibroblasts were used to investigate axonal mitochondrial trafficking and the nature of nuclear indentations. METHODS: Pathological and control iPSCs generated from patients with a low number of repeats were differentiated in striatal neurons of the brain. Mitochondrial density was measured along the axon using tubulin beta 3 co-staining in pathological and control neurons. To investigate the connection of nuclear roundness with calcium dysregulation, several calcium inhibitors were used. Proteasome system inhibition was applied to mimic premature neuronal ageing. RESULTS: We found that the mitochondrial density was approximately 7.6 ± 0.2 in neurites in control neurons but was only 5.3 ± 0.2 in mutant neurons with 40-44 CAG repeats (p-value <0.005). Neuronal ageing induced by proteasome inhibitor MG132 significantly decreased the mitochondrial density by 15% and 25% in control and mutant neurons to 6.5 ± 0.1 (p-value < 0.005) and 4.0 ± 0.3 (p-value < 0.005), respectively. Thus, for the first time, an impairment of mitochondrial trafficking in pathological neurons with endogenous mutant huntingtin was demonstrated. We found that inhibiting the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the ryanodine-receptor (RyR) or the inositol 1,4,5-trisphosphate receptor (IP3R) by specific inhibitors did not specifically affect the nuclear roundness or survival of pathological neurons differentiated from patient iPSCs. Therefore, nuclear calcium homeostasis is not directly associated with HD pathology. CONCLUSION: Identifying HD iPSCs and differentiating from them neurons provide a unique system for modelling the disease in vitro. Impairments of mitochondrial trafficking and nuclear roundness manifest long before the disease onset, while premature neuronal ageing enhances differences in mitochondrial distribution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


