Ivone U S Leong, Jonathan R Skinner, Andrew N Shelling, Donald R Love
{"title":"Expression of a Mutant kcnj2 Gene Transcript in Zebrafish.","authors":"Ivone U S Leong, Jonathan R Skinner, Andrew N Shelling, Donald R Love","doi":"10.1155/2013/324839","DOIUrl":null,"url":null,"abstract":"<p><p>Long QT 7 syndrome (LQT7, also known as Andersen-Tawil syndrome) is a rare autosomal-dominant disorder that causes cardiac arrhythmias, periodic paralysis, and dysmorphic features. Mutations in the human KCNJ2 gene, which encodes for the subunit of the potassium inwardly-rectifying channel (IK1), have been associated with the disorder. The majority of mutations are considered to be dominant-negative as mutant proteins interact to limit the function of wild type KCNJ2 proteins. Several LQT7 syndrome mouse models have been created that vary in the physiological similarity to the human disease. To complement the LQT7 mouse models, we investigated the usefulness of the zebrafish as an alternative model via a transient approach. Initial bioinformatic analysis identified the zebrafish orthologue of the human KCNJ2 gene, together with a spatial expression profile that was similar to that of human. The expression of a kcnj2-12 transcript carrying an in-frame deletion of critical amino acids identified in human studies resulted in embryos that exhibited defects in muscle development, thereby affecting movement, a decrease in jaw size, pupil-pupil distance, and signs of scoliosis. These defects correspond to some phenotypes expressed by human LQT7 patients. </p>","PeriodicalId":89785,"journal":{"name":"ISRN molecular biology","volume":"2013 ","pages":"324839"},"PeriodicalIF":0.0000,"publicationDate":"2013-11-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4890933/pdf/","citationCount":"11","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN molecular biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/324839","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 11
Abstract
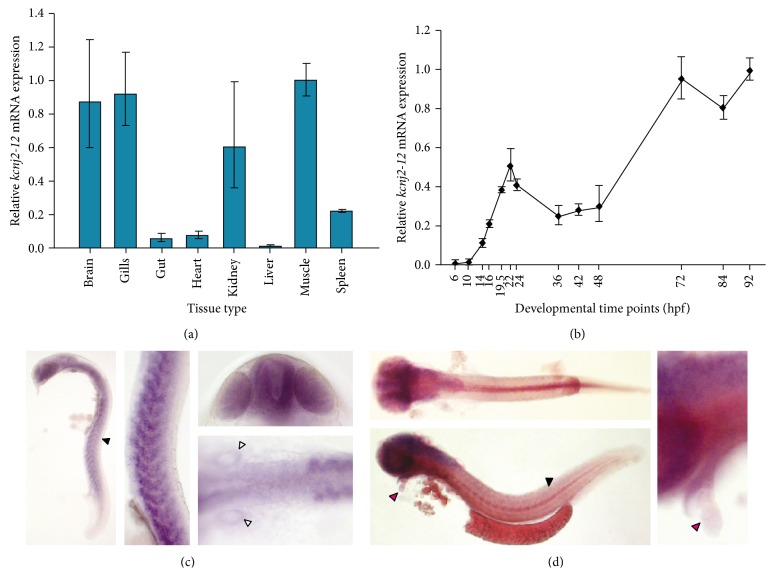
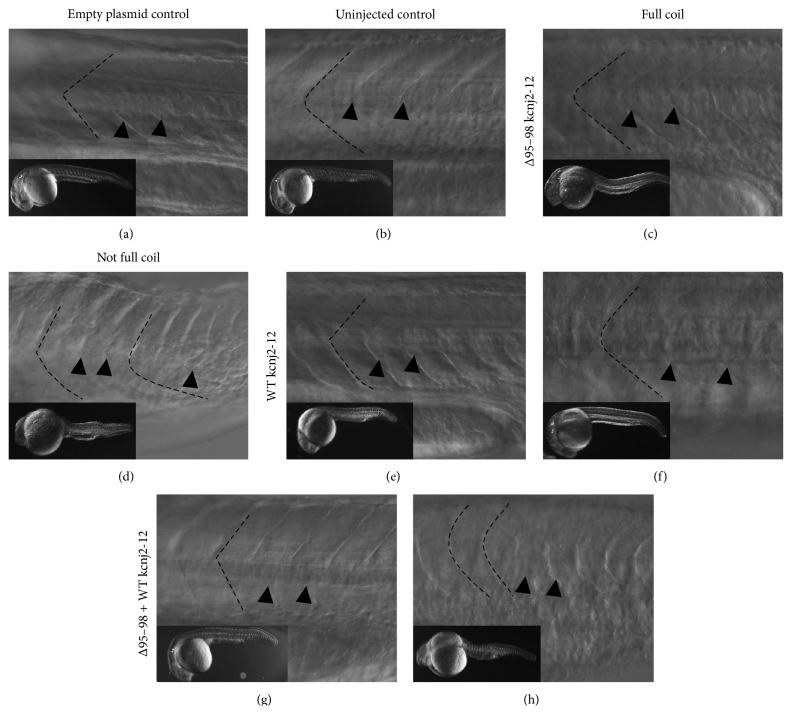
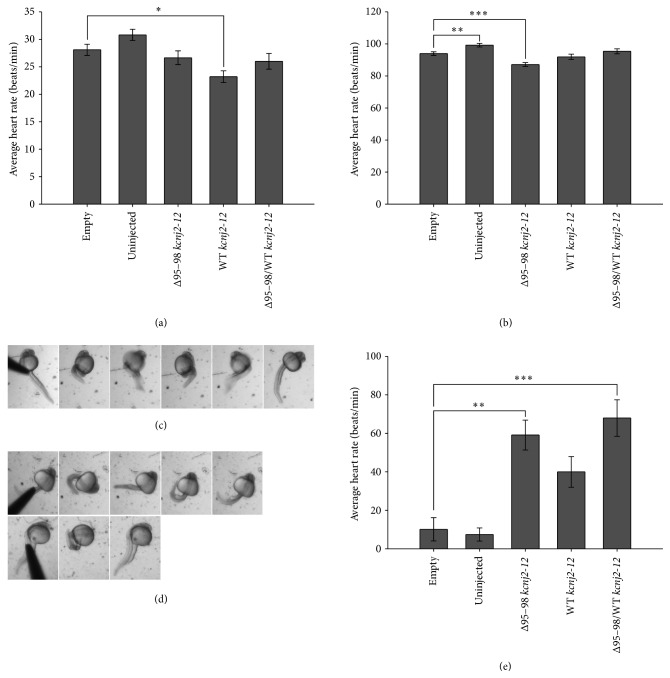
Long QT 7 syndrome (LQT7, also known as Andersen-Tawil syndrome) is a rare autosomal-dominant disorder that causes cardiac arrhythmias, periodic paralysis, and dysmorphic features. Mutations in the human KCNJ2 gene, which encodes for the subunit of the potassium inwardly-rectifying channel (IK1), have been associated with the disorder. The majority of mutations are considered to be dominant-negative as mutant proteins interact to limit the function of wild type KCNJ2 proteins. Several LQT7 syndrome mouse models have been created that vary in the physiological similarity to the human disease. To complement the LQT7 mouse models, we investigated the usefulness of the zebrafish as an alternative model via a transient approach. Initial bioinformatic analysis identified the zebrafish orthologue of the human KCNJ2 gene, together with a spatial expression profile that was similar to that of human. The expression of a kcnj2-12 transcript carrying an in-frame deletion of critical amino acids identified in human studies resulted in embryos that exhibited defects in muscle development, thereby affecting movement, a decrease in jaw size, pupil-pupil distance, and signs of scoliosis. These defects correspond to some phenotypes expressed by human LQT7 patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


