{"title":"COVID-19 Whole-Genome Resequencing with Redundant Tiling PCR and Subtract-Based Amplicon Normalization Successfully Characterized SARS-CoV-2 Variants in Clinical Specimens.","authors":"Tatsuki Sugi, Mizanur Rahman, Rummana Rahim, Abu Hasan, Naoko Kawai, Kyoko Hayashida, Junya Yamagishi","doi":"10.1155/2022/2109641","DOIUrl":null,"url":null,"abstract":"<p><p>With an increasing number of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) sequences gathered worldwide, we recognize that deletion mutants and nucleotide substitutions that may affect whole-genome sequencing are accumulating. Here, we propose an additional strategy for tiling PCR for whole-genome resequencing, which can make the pipeline robust for mutations at the primer annealing site by a redundant amplicon scheme. We further demonstrated that subtracting overrepresented amplicons from the multiplex PCR products reduced the bias of the next-generation sequencing (NGS) library, resulting in decreasing required sequencing reads per sample. We applied this sequencing strategy to clinical specimens collected in Bangladesh. More than 80% out of the 304 samples were successfully sequenced. Less than 5% were ambiguous nucleotides, and several known variants were detected. With the additional strategies presented here, we believe that whole-genome resequencing of SARS-CoV-2 from clinical samples can be optimized.</p>","PeriodicalId":39128,"journal":{"name":"Interdisciplinary Perspectives on Infectious Diseases","volume":" ","pages":"2109641"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9534710/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Interdisciplinary Perspectives on Infectious Diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2022/2109641","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Immunology and Microbiology","Score":null,"Total":0}
引用次数: 0
Abstract
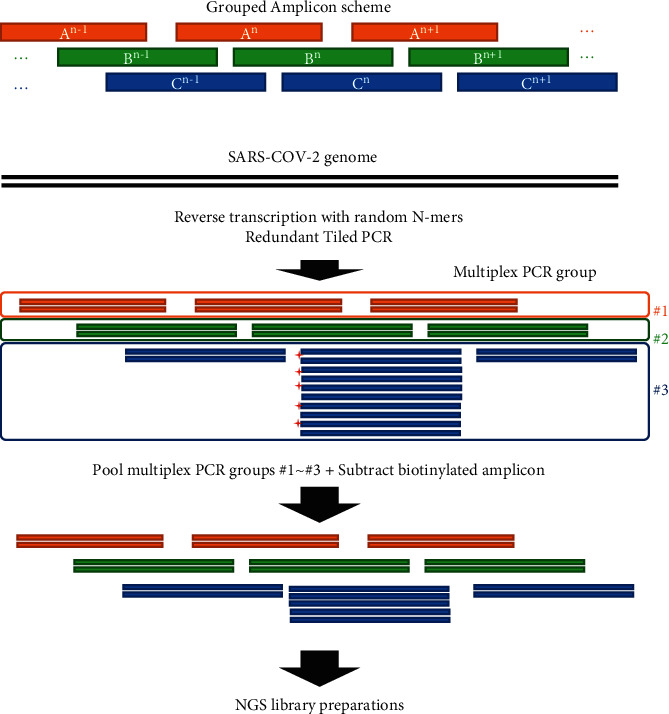
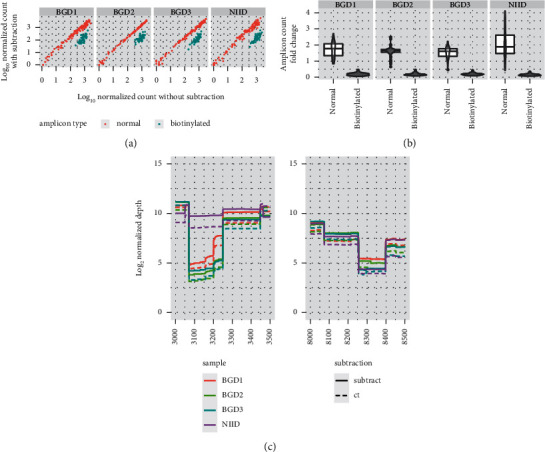
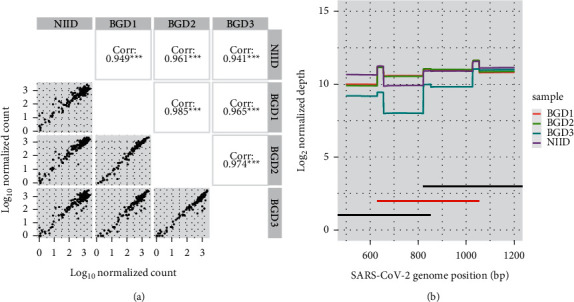
With an increasing number of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) sequences gathered worldwide, we recognize that deletion mutants and nucleotide substitutions that may affect whole-genome sequencing are accumulating. Here, we propose an additional strategy for tiling PCR for whole-genome resequencing, which can make the pipeline robust for mutations at the primer annealing site by a redundant amplicon scheme. We further demonstrated that subtracting overrepresented amplicons from the multiplex PCR products reduced the bias of the next-generation sequencing (NGS) library, resulting in decreasing required sequencing reads per sample. We applied this sequencing strategy to clinical specimens collected in Bangladesh. More than 80% out of the 304 samples were successfully sequenced. Less than 5% were ambiguous nucleotides, and several known variants were detected. With the additional strategies presented here, we believe that whole-genome resequencing of SARS-CoV-2 from clinical samples can be optimized.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


