{"title":"In vitro-in vivo correlation evaluation of generic alfuzosin modified release tablets.","authors":"Utpal Kumar Sanki, Badal Kumar Mandal","doi":"10.5402/2012/813836","DOIUrl":null,"url":null,"abstract":"<p><p>Alfuzosin, a selective alpha-1a antagonistis is the most recently approved AARAS, with limited cardiac toxicity and exclusively used for lower urinary tract syndromes (LUTS). In order to reduce pill burden and better patient compliance modified release (MR) formulations have been developed. Alfuzosin MR tablet was developed by the use of hot-melt granulation techniques using mono- and diglycerides as rate controlling membranes to minimize health care cost and uses of costly excipients. The other purpose of the study was to evaluate in vitro-in vivo performance of the scale up batch in healthy human subjects for commercialization. The blend uniformity (mean ± RSD%), assay, cumulative percent dissolution at 24 h, hardness, and friability of the biobatch were 100.2 ± 0.05%, 100.43 ± 0.023%, 93.98%, 4.5 kg, 5 min, and 0.08%, respectively. The in vivo pharmacokinetic parameters under fasting conditions between test and reference formulations (Uroxatral 10 mg extended release tablets) were comparable. The 90% CI, geometric mean ratio (%) and power of C max, AUCT, and AUCI of the fasting study for the test and reference formulation were 99.03% to 122.78%, 109%, 0.998; 92.94% to 116.71%, 104%, 1; 98.17% to 124.01%, 110% 1, respectively. The scale up biobatch showed negligible difference in in vitro properties with respect to the pilot batch. The formulation developed with these agents was safe to use as there were no serious adverse events developed during the conduction of the clinical trial on the healthy subjects. Furthermore, the developed formulation was bioequivalent with respect to rate and extends of absorption to the reference formulation.</p>","PeriodicalId":14674,"journal":{"name":"ISRN Toxicology","volume":"2012 ","pages":"813836"},"PeriodicalIF":0.0000,"publicationDate":"2012-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.5402/2012/813836","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN Toxicology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5402/2012/813836","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/1/1 0:00:00","PubModel":"Print","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
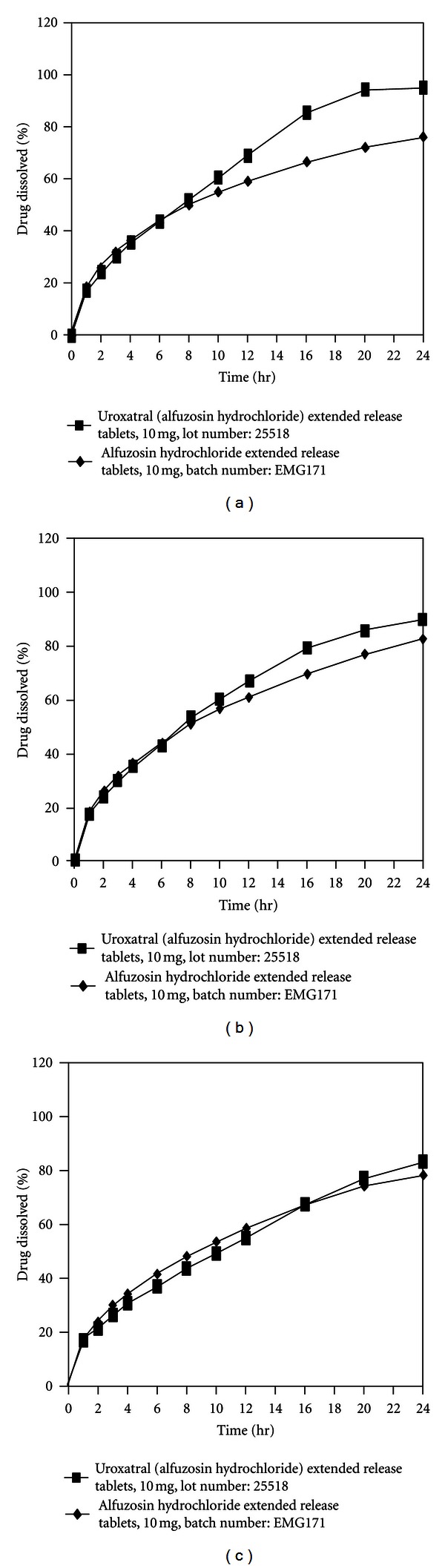
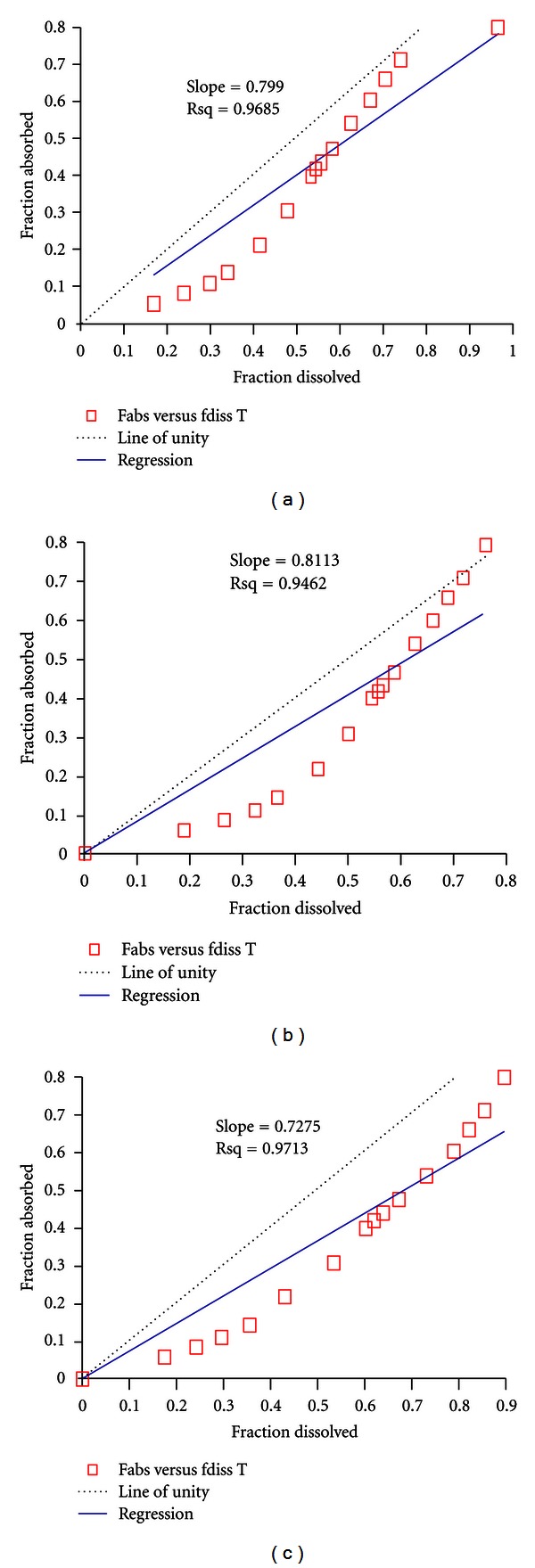
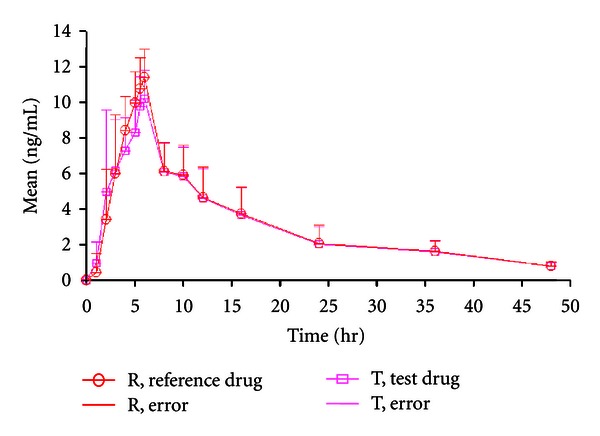
Alfuzosin, a selective alpha-1a antagonistis is the most recently approved AARAS, with limited cardiac toxicity and exclusively used for lower urinary tract syndromes (LUTS). In order to reduce pill burden and better patient compliance modified release (MR) formulations have been developed. Alfuzosin MR tablet was developed by the use of hot-melt granulation techniques using mono- and diglycerides as rate controlling membranes to minimize health care cost and uses of costly excipients. The other purpose of the study was to evaluate in vitro-in vivo performance of the scale up batch in healthy human subjects for commercialization. The blend uniformity (mean ± RSD%), assay, cumulative percent dissolution at 24 h, hardness, and friability of the biobatch were 100.2 ± 0.05%, 100.43 ± 0.023%, 93.98%, 4.5 kg, 5 min, and 0.08%, respectively. The in vivo pharmacokinetic parameters under fasting conditions between test and reference formulations (Uroxatral 10 mg extended release tablets) were comparable. The 90% CI, geometric mean ratio (%) and power of C max, AUCT, and AUCI of the fasting study for the test and reference formulation were 99.03% to 122.78%, 109%, 0.998; 92.94% to 116.71%, 104%, 1; 98.17% to 124.01%, 110% 1, respectively. The scale up biobatch showed negligible difference in in vitro properties with respect to the pilot batch. The formulation developed with these agents was safe to use as there were no serious adverse events developed during the conduction of the clinical trial on the healthy subjects. Furthermore, the developed formulation was bioequivalent with respect to rate and extends of absorption to the reference formulation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


