{"title":"Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus.","authors":"Zhongmin Alex Ma, Zhengshan Zhao, John Turk","doi":"10.1155/2012/703538","DOIUrl":null,"url":null,"abstract":"<p><p>Type 2 diabetes mellitus (T2DM) is the most common human endocrine disease and is characterized by peripheral insulin resistance and pancreatic islet β-cell failure. Accumulating evidence indicates that mitochondrial dysfunction is a central contributor to β-cell failure in the evolution of T2DM. As reviewed elsewhere, reactive oxygen species (ROS) produced by β-cell mitochondria as a result of metabolic stress activate several stress-response pathways. This paper focuses on mechanisms whereby ROS affect mitochondrial structure and function and lead to β-cell failure. ROS activate UCP2, which results in proton leak across the mitochondrial inner membrane, and this leads to reduced β-cell ATP synthesis and content, which is a critical parameter in regulating glucose-stimulated insulin secretion. In addition, ROS oxidize polyunsaturated fatty acids in mitochondrial cardiolipin and other phospholipids, and this impairs membrane integrity and leads to cytochrome c release into cytosol and apoptosis. Group VIA phospholipase A₂ (iPLA₂β) appears to be a component of a mechanism for repairing mitochondrial phospholipids that contain oxidized fatty acid substituents, and genetic or acquired iPLA₂β-deficiency increases β-cell mitochondrial susceptibility to injury from ROS and predisposes to developing T2DM. Interventions that attenuate ROS effects on β-cell mitochondrial phospholipids might prevent or retard development of T2DM.</p>","PeriodicalId":12109,"journal":{"name":"Experimental Diabetes Research","volume":"2012 ","pages":"703538"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3216264/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Experimental Diabetes Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2012/703538","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2011/11/9 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
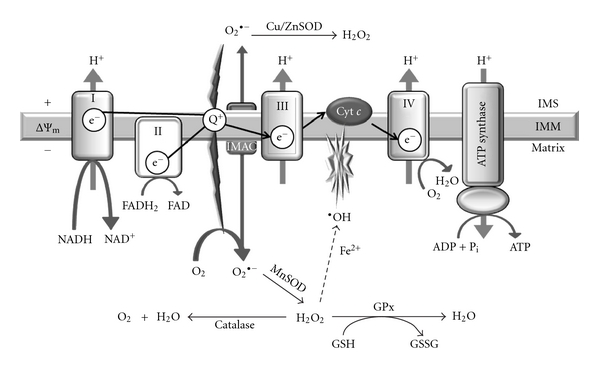
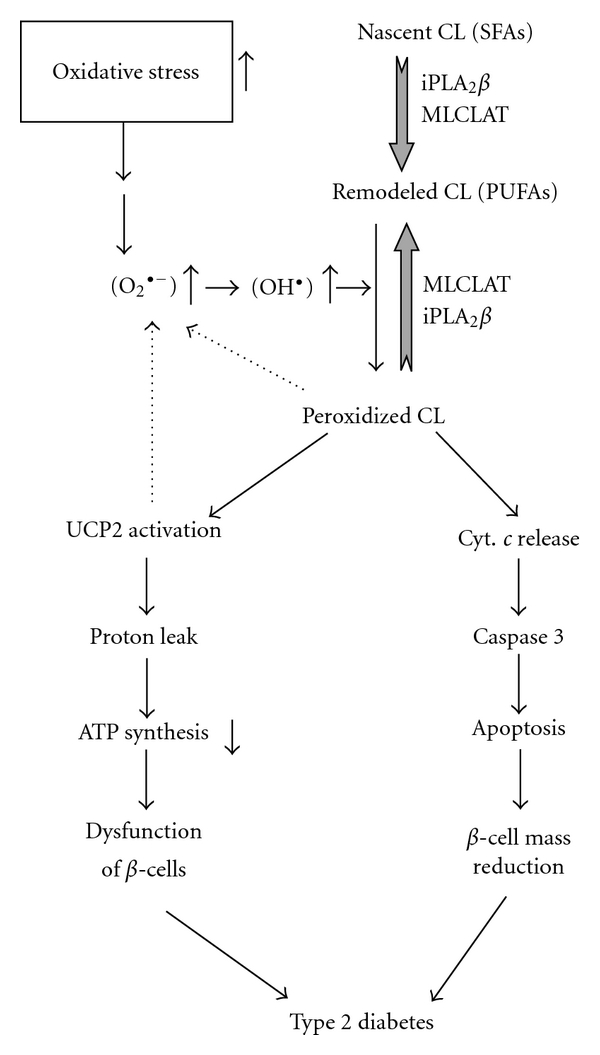
Type 2 diabetes mellitus (T2DM) is the most common human endocrine disease and is characterized by peripheral insulin resistance and pancreatic islet β-cell failure. Accumulating evidence indicates that mitochondrial dysfunction is a central contributor to β-cell failure in the evolution of T2DM. As reviewed elsewhere, reactive oxygen species (ROS) produced by β-cell mitochondria as a result of metabolic stress activate several stress-response pathways. This paper focuses on mechanisms whereby ROS affect mitochondrial structure and function and lead to β-cell failure. ROS activate UCP2, which results in proton leak across the mitochondrial inner membrane, and this leads to reduced β-cell ATP synthesis and content, which is a critical parameter in regulating glucose-stimulated insulin secretion. In addition, ROS oxidize polyunsaturated fatty acids in mitochondrial cardiolipin and other phospholipids, and this impairs membrane integrity and leads to cytochrome c release into cytosol and apoptosis. Group VIA phospholipase A₂ (iPLA₂β) appears to be a component of a mechanism for repairing mitochondrial phospholipids that contain oxidized fatty acid substituents, and genetic or acquired iPLA₂β-deficiency increases β-cell mitochondrial susceptibility to injury from ROS and predisposes to developing T2DM. Interventions that attenuate ROS effects on β-cell mitochondrial phospholipids might prevent or retard development of T2DM.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


