Systematic comparison of crystal and NMR protein structures deposited in the protein data bank.
Q3 Biochemistry, Genetics and Molecular Biology
引用次数: 67
Abstract
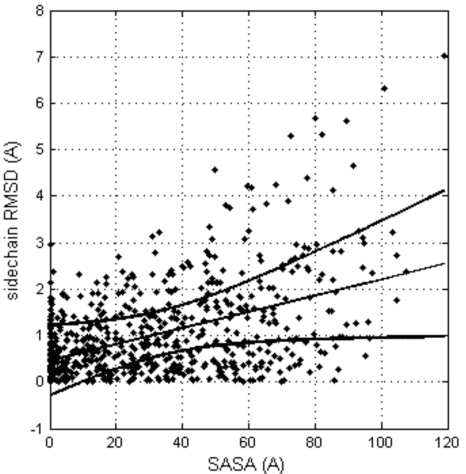
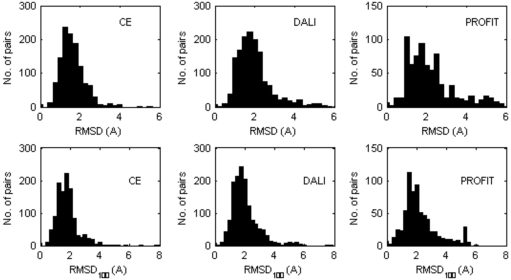
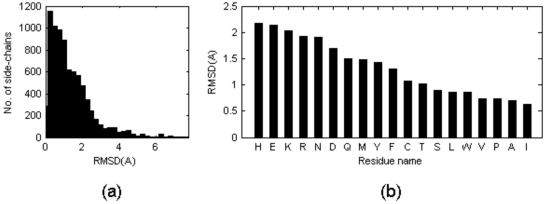
Nearly all the macromolecular three-dimensional structures deposited in Protein Data Bank were determined by either crystallographic (X-ray) or Nuclear Magnetic Resonance (NMR) spectroscopic methods. This paper reports a systematic comparison of the crystallographic and NMR results deposited in the files of the Protein Data Bank, in order to find out to which extent these information can be aggregated in bioinformatics. A non-redundant data set containing 109 NMR – X-ray structure pairs of nearly identical proteins was derived from the Protein Data Bank. A series of comparisons were performed by focusing the attention towards both global features and local details. It was observed that: (1) the RMDS values between NMR and crystal structures range from about 1.5 Å to about 2.5 Å; (2) the correlation between conformational deviations and residue type reveals that hydrophobic amino acids are more similar in crystal and NMR structures than hydrophilic amino acids; (3) the correlation between solvent accessibility of the residues and their conformational variability in solid state and in solution is relatively modest (correlation coefficient = 0.462); (4) beta strands on average match better between NMR and crystal structures than helices and loops; (5) conformational differences between loops are independent of crystal packing interactions in the solid state; (6) very seldom, side chains buried in the protein interior are observed to adopt different orientations in the solid state and in solution.
系统的比较晶体和核磁共振蛋白质结构沉积在蛋白质数据库。
几乎所有沉积在蛋白质数据库中的大分子三维结构都是通过晶体学(x射线)或核磁共振(NMR)光谱方法确定的。本文报道了一个系统的比较结晶学和核磁共振结果存储在蛋白质数据库的文件,以找出在何种程度上这些信息可以聚集在生物信息学。从蛋白质数据库中获得了包含109个核磁共振- x射线结构对的非冗余数据集。通过将注意力集中在全局特征和局部细节上,进行了一系列的比较。结果表明:(1)核磁共振与晶体结构之间的RMDS值在1.5 Å ~ 2.5 Å之间;(2)构象偏差与残基类型的相关性表明,疏水氨基酸在晶体和核磁共振结构上比亲水性氨基酸更相似;(3)残基的溶剂可溶性与其固溶构象变异性的相关性较弱(相关系数= 0.462);(4) β链核磁共振与晶体结构的平均匹配优于螺旋和环;(5)环之间的构象差异与固体中晶体堆积的相互作用无关;(6)很少观察到埋在蛋白质内部的侧链在固态和溶液中采用不同的取向。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Open Biochemistry Journal
Biochemistry, Genetics and Molecular Biology-Biochemistry, Genetics and Molecular Biology (all)
CiteScore
1.50
自引率
0.00%
发文量
5
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


