Computational Prediction of Stacking Mode in Conductive Two-Dimensional Metal–Organic Frameworks: An Exploration of Chemical and Electrical Property Changes
Mingyu Jeon, Minhyuk Kim, Joon-Seok Lee, Honghui Kim, Seon-Jin Choi, Hoi Ri Moon and Jihan Kim*,
{"title":"Computational Prediction of Stacking Mode in Conductive Two-Dimensional Metal–Organic Frameworks: An Exploration of Chemical and Electrical Property Changes","authors":"Mingyu Jeon, Minhyuk Kim, Joon-Seok Lee, Honghui Kim, Seon-Jin Choi, Hoi Ri Moon and Jihan Kim*, ","doi":"10.1021/acssensors.3c00715","DOIUrl":null,"url":null,"abstract":"<p >Conductive two-dimensional metal–organic frameworks (2D MOFs) have attracted interest as they induce strong charge delocalization and improve charge carrier mobility and concentration. However, characterizing their stacking mode depends on expensive and time-consuming experimental measurements. Here, we construct a potential energy surface (PES) map database for 36 2D MOFs using density functional theory (DFT) for the experimentally synthesized and non-synthesized 2D MOFs to predict their stacking mode. The DFT PES results successfully predict the experimentally synthesized stacking mode with an accuracy of 92.9% and explain the coexistence mechanism of dual stacking modes in a single compound. Furthermore, we analyze the chemical (i.e., host–guest interaction) and electrical (i.e., electronic structure) property changes affected by stacking mode. The DFT results show that the host–guest interaction can be enhanced by the transition from AA to AB stacking, taking H<sub>2</sub>S gas as a case study. The electronic band structure calculation confirms that as AB stacking displacement increases, the in-plane charge transport pathway is reduced while the out-of-plane charge transport pathway is maintained or even increased. These results indicate that there is a trade-off between chemical and electrical properties in accordance with the stacking mode.</p>","PeriodicalId":24,"journal":{"name":"ACS Sensors","volume":"8 8","pages":"3068–3075"},"PeriodicalIF":9.1000,"publicationDate":"2023-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Sensors","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acssensors.3c00715","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, ANALYTICAL","Score":null,"Total":0}
引用次数: 3
Abstract
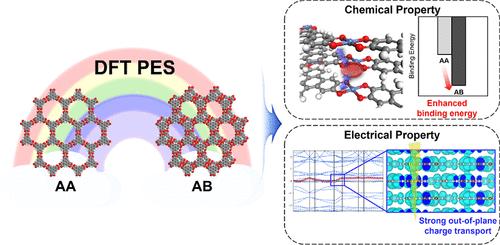
Conductive two-dimensional metal–organic frameworks (2D MOFs) have attracted interest as they induce strong charge delocalization and improve charge carrier mobility and concentration. However, characterizing their stacking mode depends on expensive and time-consuming experimental measurements. Here, we construct a potential energy surface (PES) map database for 36 2D MOFs using density functional theory (DFT) for the experimentally synthesized and non-synthesized 2D MOFs to predict their stacking mode. The DFT PES results successfully predict the experimentally synthesized stacking mode with an accuracy of 92.9% and explain the coexistence mechanism of dual stacking modes in a single compound. Furthermore, we analyze the chemical (i.e., host–guest interaction) and electrical (i.e., electronic structure) property changes affected by stacking mode. The DFT results show that the host–guest interaction can be enhanced by the transition from AA to AB stacking, taking H2S gas as a case study. The electronic band structure calculation confirms that as AB stacking displacement increases, the in-plane charge transport pathway is reduced while the out-of-plane charge transport pathway is maintained or even increased. These results indicate that there is a trade-off between chemical and electrical properties in accordance with the stacking mode.
期刊介绍:
ACS Sensors is a peer-reviewed research journal that focuses on the dissemination of new and original knowledge in the field of sensor science, particularly those that selectively sense chemical or biological species or processes. The journal covers a broad range of topics, including but not limited to biosensors, chemical sensors, gas sensors, intracellular sensors, single molecule sensors, cell chips, and microfluidic devices. It aims to publish articles that address conceptual advances in sensing technology applicable to various types of analytes or application papers that report on the use of existing sensing concepts in new ways or for new analytes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


