SlaKoNet: A Unified Slater-Koster Tight-Binding Framework Using Neural Network Infrastructure for the Periodic Table
IF 4.6
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
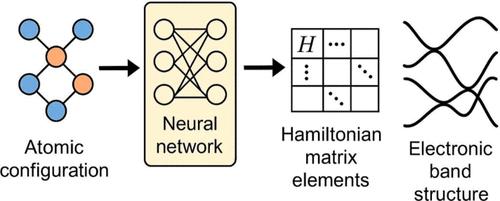
Accurate and efficient prediction of electronic band structures is essential for designing materials with targeted properties. However, existing machine learning models often lack universality and struggle to predict detailed electronic structures, while traditional tight-binding models based on the Slater-Koster (SK) formalism suffer from (i) limited transferability, (ii) the need for manual parametrization, and (iii) training on low-fidelity electronic structure data. To address these challenges, I introduce SlaKoNet, a parameter optimization framework that learns SK-based Hamiltonian matrix elements across 65 elements of the periodic table using automatic differentiation. SlaKoNet is trained on density functional theory data from the JARVIS-DFT database using the Tran-Blaha modified Becke-Johnson (TBmBJ) functional, encompassing over 20000 materials. The framework achieves a mean absolute error (MAE) of 0.74 eV for bandgap predictions against experimental data, representing a reasonable improvement over standard GGA functionals (MAE = 1.14 eV) while preserving the computational advantages and physical interpretability of tight-binding methods. SlaKoNet demonstrates promising scalability with up to 8.4× speedup on GPUs, enabling rapid electronic structure screening for materials discovery. SlaKoNet is publicly available at the Web site https://github.com/atomgptlab/slakonet.
SlaKoNet:使用神经网络基础设施的元素周期表统一的Slater-Koster紧密绑定框架
准确有效地预测电子能带结构对于设计具有目标性能的材料至关重要。然而,现有的机器学习模型往往缺乏通用性,难以预测详细的电子结构,而基于Slater-Koster (SK)形式主义的传统紧密绑定模型存在以下问题:(i)有限的可移植性,(ii)需要手动参数化,以及(iii)在低保真电子结构数据上进行训练。为了解决这些挑战,我介绍了SlaKoNet,这是一个参数优化框架,它使用自动微分学习元素周期表中65个元素的基于sk的哈密顿矩阵元素。SlaKoNet使用trans - blaha修改的Becke-Johnson (TBmBJ)泛函对来自JARVIS-DFT数据库的密度泛函理论数据进行训练,包含超过20000种材料。该框架对实验数据的带隙预测的平均绝对误差(MAE)为0.74 eV,比标准GGA函数(MAE = 1.14 eV)有了合理的改进,同时保留了紧密结合方法的计算优势和物理可解释性。SlaKoNet在gpu上具有高达8.4倍的加速,具有良好的可扩展性,可实现材料发现的快速电子结构筛选。SlaKoNet在网站https://github.com/atomgptlab/slakonet上公开提供。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


