{"title":"Excited-State Densities from Time-Dependent Density Functional Response Theory.","authors":"Anna Baranova, Neepa T Maitra","doi":"10.1021/acs.jctc.5c00909","DOIUrl":null,"url":null,"abstract":"<p><p>While the variational principle for excited-state energies leads to a route to obtaining excited-state densities from time-dependent density functional theory, relatively little attention has been paid to the quality of the resulting densities in real space obtained with different exchange-correlation functional approximations or how nonadiabatic approximations developed for energies of states of double-excitation character perform for their densities. Here we derive an expression directly in real space for the excited-state density, which includes the case of nonadiabatic kernels and consequently is able, for the first time, to yield densities of states of double-excitation character. Under some well-defined simplifications, we compare the performance of the local-density approximation and exact-exchange approximation, which are in a sense at the opposite extremes of the fundamental functional approximations, on local and charge-transfer excitations in one-dimensional model systems and show that the dressed Time-Dependent Density Functional Theory (TDDFT) approach gives good densities of double excitations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c00909","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
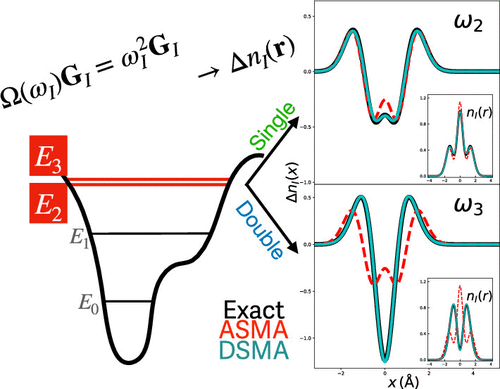
While the variational principle for excited-state energies leads to a route to obtaining excited-state densities from time-dependent density functional theory, relatively little attention has been paid to the quality of the resulting densities in real space obtained with different exchange-correlation functional approximations or how nonadiabatic approximations developed for energies of states of double-excitation character perform for their densities. Here we derive an expression directly in real space for the excited-state density, which includes the case of nonadiabatic kernels and consequently is able, for the first time, to yield densities of states of double-excitation character. Under some well-defined simplifications, we compare the performance of the local-density approximation and exact-exchange approximation, which are in a sense at the opposite extremes of the fundamental functional approximations, on local and charge-transfer excitations in one-dimensional model systems and show that the dressed Time-Dependent Density Functional Theory (TDDFT) approach gives good densities of double excitations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


