DFT-based investigation of polymer components for concrete impregnation: Electronic and structural insights
IF 4.9
3区 材料科学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
This study presents a systematic density functional theory (DFT) investigation of the structural, electronic, and vibrational properties of polymer components used in concrete impregnation, namely styrene, divinyl benzene, and benzoyl peroxide. The molecular geometries of monomers and their oligomeric structures were optimized, and their electronic descriptors were analyzed to provide insights into stability and reactivity. The calculated HOMO–LUMO energy gaps indicated semiconducting behavior for the monomers, while a significant band gap reduction was observed with increasing polymer chain length, suggesting enhanced charge-transfer ability and optical activity during polymerization. Vibrational frequency analysis confirmed the characteristic modes of functional groups responsible for polymerization. In addition, global reactivity descriptors such as hardness, softness, and electrophilicity were evaluated to elucidate the trends associated with molecular growth. The findings highlight the strong correlation between chain length and electronic stability, and provide predictive insights into the performance of polymer–concrete composites at the molecular level. This theoretical framework complements experimental studies and may guide the design of more durable polymer-modified concrete systems.
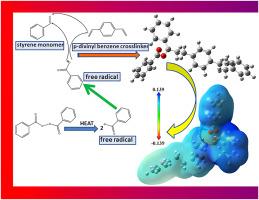
基于dft的混凝土浸渍聚合物组分研究:电子和结构见解
本研究提出了一个系统的密度泛函理论(DFT)研究在混凝土浸渍中使用的聚合物组分,即苯乙烯、二乙烯基苯和过氧化苯甲酰的结构、电子和振动特性。优化了单体的分子几何形状及其寡聚结构,并分析了它们的电子描述符,以提供稳定性和反应性的见解。计算得到的HOMO-LUMO能隙表明单体具有半导体性质,而随着聚合物链长度的增加,带隙明显减小,表明聚合过程中的电荷转移能力和光学活性增强。振动频率分析证实了聚合官能团的特征模式。此外,还评估了整体反应性描述符,如硬度,柔软性和亲电性,以阐明与分子生长相关的趋势。研究结果强调了链长与电子稳定性之间的强相关性,并在分子水平上为聚合物-混凝土复合材料的性能提供了预测性见解。这一理论框架补充了实验研究,并可能指导更耐用的聚合物改性混凝土体系的设计。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
7.80
自引率
2.50%
发文量
605
审稿时长
40 days
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


