Ab-Initio investigation of the structural, elastic, electronic, magnetic and optical properties of Mn-doped ZnSe wurtzite: Application in solar cells
IF 4.9
3区 材料科学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
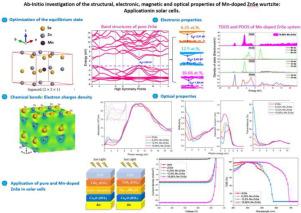
This research proposes a comprehensive ab initio study of the structural, electronic, mechanical, magnetic, and optical properties of pure ZnSe and Mn-doped ZnSe at different concentrations using density functional theory (DFT) according to the GGA-PBE+U approximation. Examination of the electronic band structure reveals that pure ZnSe exhibits a direct band gap of 2.69 eV at the point, which agrees well with experimental data. Our results demonstrate that the integration of Mn systematically modifies the fundamental characteristics of ZnSe. Structurally, the addition of Mn causes a lattice expansion: for a Mn content of 16.66%, the lattice parameters extend to a = 4.034 Å and c = 6.610 Å, which indicates a lattice expansion while preserving mechanical stability (Young’s modulus 137.7 GPa, Poisson’s ratio 0.29, B/G 2.03). Electronically, the energy gap gradually decreases from 2.51 eV at 6.25% Mn to 1.89 eV at 16.66% Mn, due to Mn 3d-Se 4s hybridization, which is confirmed by density of states and charge density analyses. In terms of magnetism, Mn elicits ferromagnetism with local magnetic moments of about 5 B per Mn atom; the ferromagnetic phase is energetically preferential ( = 0.0482 eV/supercell), whereas the non-magnetic phase is significantly less stable ( = 0.8003 eV). The exchange constants ( 0.19 eV; = 1.3–2.0 eV) indicate predominant ferromagnetic interactions. Mn doping significantly enhances optical properties: absorption in the visible and infrared regions increases, reflectivity increases in the infrared and low-visible region while decreasing in the UV, and transmission increases in the IR region. These changes show a quantitative correlation with Mn concentration, illustrating tunable optoelectronic behavior. Photovoltaic simulations also show optimal device performance with 16.66% Mn doping, displaying a Voc of 0.8849 V, a Jsc of 16.90 mA/cm2, a fill factor FF of 87.06%, and a PCE energy conversion efficiency of 13.03%, more than double compared to pure ZnSe (6.44%). The external quantum efficiency reaches 100% in the 400–550 nm range and remains high up to 690 nm, with an optimal absorber thickness of approximately . In summary, our research quantitatively establishes the link between structure, magnetism, optical response and photovoltaic efficiency, highlighting Mn-doped ZnSe as a multifunctional material with promising applications in spintronics, optoelectronics, infrared detectors and highly efficient solar cells.
mn掺杂ZnSe纤锌矿结构、弹性、电子、磁性和光学性质的Ab-Initio研究:在太阳能电池中的应用
本研究根据GGA-PBE+U近似,利用密度泛函理论(DFT)对不同浓度纯ZnSe和mn掺杂ZnSe的结构、电子、机械、磁性和光学性质进行了全面的从头算研究。电子能带结构检测表明,纯ZnSe在Γ点的直接带隙为2.69 eV,与实验数据吻合较好。我们的研究结果表明,Mn的集成系统地改变了ZnSe的基本特性。在结构上,Mn的加入引起晶格膨胀,当Mn含量为16.66%时,晶格参数扩展到a = 4.034 Å和c = 6.610 Å,表明晶格膨胀同时保持了力学稳定性(杨氏模量≈137.7 GPa,泊松比ν≈0.29,B/G≈2.03)。从电子角度看,由于Mn 3d-Se - 4s杂化作用,能隙从6.25% Mn时的2.51 eV逐渐减小到16.66% Mn时的1.89 eV,态密度和电荷密度分析证实了这一点。在磁性方面,Mn原子具有铁磁性,其局部磁矩约为5 μB;铁磁相能量优先(ΔE1 = 0.0482 eV/超级单体),而非磁相能量不稳定(ΔE1 = 0.8003 eV)。交换常数(N0α≈0.19 eV; N0β = 1.3 ~ 2.0 eV)表明主要的铁磁相互作用。Mn掺杂显著增强了光学性能:可见光和红外区的吸收率增加,红外和低可见光区的反射率增加,紫外区的反射率降低,红外区的透射率增加。这些变化显示出与Mn浓度的定量相关性,说明了可调谐的光电行为。光伏模拟结果表明,当Mn掺杂率为16.66%时,器件性能最佳,Voc为0.8849 V, Jsc为16.90 mA/cm2,填充系数FF为87.06%,PCE能量转换效率为13.03%,是纯ZnSe(6.44%)的两倍多。在400-550 nm范围内,外量子效率达到100%,在690 nm范围内保持高效率,最佳吸收层厚度约为1.8μm。总之,我们的研究定量地建立了结构、磁性、光学响应和光伏效率之间的联系,突出了mn掺杂ZnSe作为一种多功能材料在自旋电子学、光电子学、红外探测器和高效太阳能电池方面的应用前景。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
7.80
自引率
2.50%
发文量
605
审稿时长
40 days
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


