Achilleas Betsikos, Eleni Paschou, Virginia Geladari, Evanthia Gazouni, Nikolaos Sabanis
{"title":"An Elusive Diagnosis of Autosomal Dominant Alport Syndrome: Genomic Sequencing Is a Game Changer.","authors":"Achilleas Betsikos, Eleni Paschou, Virginia Geladari, Evanthia Gazouni, Nikolaos Sabanis","doi":"10.7759/cureus.94077","DOIUrl":null,"url":null,"abstract":"<p><p>We report the case of a 55-year-old woman with chronic asymptomatic hematuria and a longstanding diagnosis of thin basement membrane disease, presenting with worsening hypertension and a significant degree of proteinuria progressing to chronic kidney disease. Genetic sequencing identified a heterozygous pathogenic variant in the COL4A4 gene (c.1321_1369+3del), a 52-base pair deletion that disrupts normal ribonucleic acid (RNA) splicing in the exon 20/intron 20 junction. In silico analysis predicted the complete loss of the canonical splice site, leading to improper splicing and the dysfunction of the α4 chain of type-IV collagen. Genetic testing in the family confirmed the presence of the same variant in two additional generations. To our knowledge, this is the first reported family in which the c.1321_1369+3del mutation is the sole cause of autosomal dominant Alport syndrome.</p>","PeriodicalId":93960,"journal":{"name":"Cureus","volume":"17 10","pages":"e94077"},"PeriodicalIF":1.3000,"publicationDate":"2025-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12504784/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cureus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.7759/cureus.94077","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/10/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
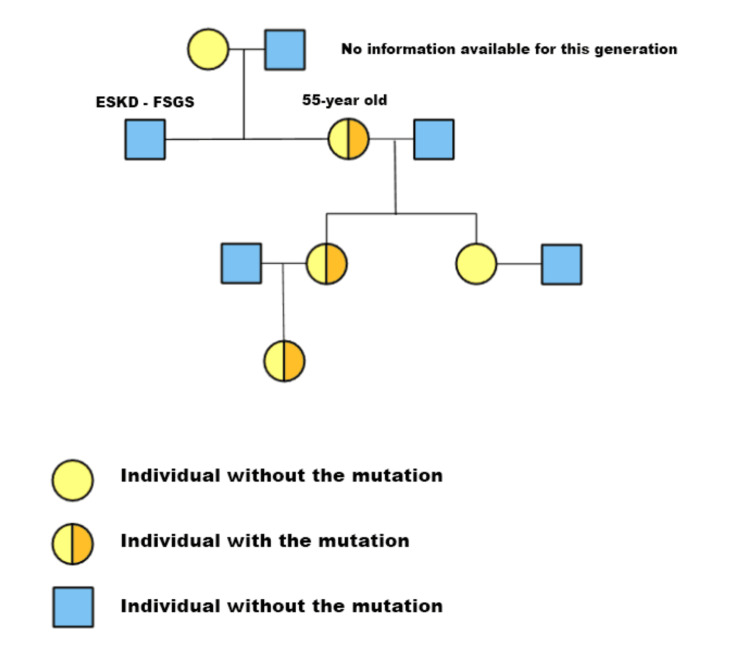
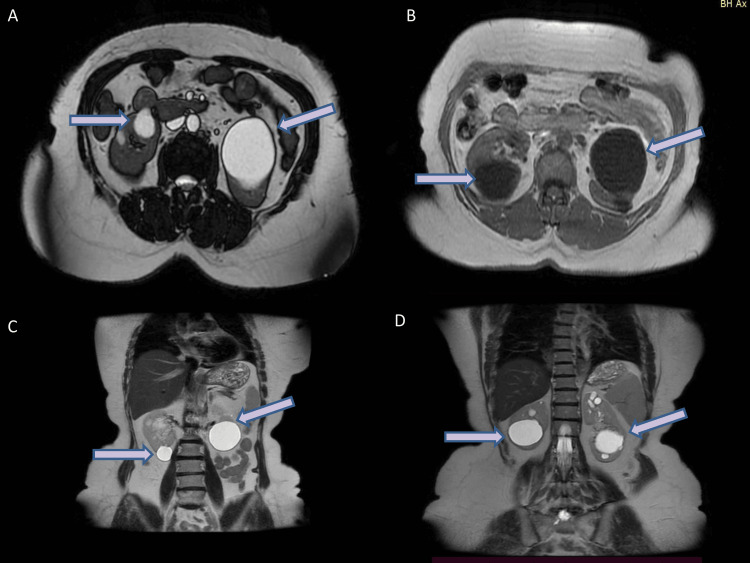
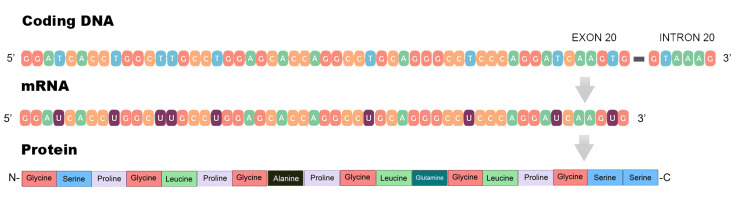
We report the case of a 55-year-old woman with chronic asymptomatic hematuria and a longstanding diagnosis of thin basement membrane disease, presenting with worsening hypertension and a significant degree of proteinuria progressing to chronic kidney disease. Genetic sequencing identified a heterozygous pathogenic variant in the COL4A4 gene (c.1321_1369+3del), a 52-base pair deletion that disrupts normal ribonucleic acid (RNA) splicing in the exon 20/intron 20 junction. In silico analysis predicted the complete loss of the canonical splice site, leading to improper splicing and the dysfunction of the α4 chain of type-IV collagen. Genetic testing in the family confirmed the presence of the same variant in two additional generations. To our knowledge, this is the first reported family in which the c.1321_1369+3del mutation is the sole cause of autosomal dominant Alport syndrome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


