Nicholas Casetti, Dylan Anstine, Olexandr Isayev, Connor W Coley
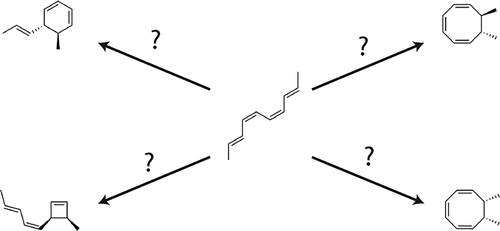
{"title":"Anticipating the Selectivity of Intramolecular Cyclization Reaction Pathways with Neural Network Potentials.","authors":"Nicholas Casetti, Dylan Anstine, Olexandr Isayev, Connor W Coley","doi":"10.1021/acs.jctc.5c01161","DOIUrl":null,"url":null,"abstract":"<p><p>Reaction mechanism search tools have demonstrated the ability to provide insights into likely products and rate-limiting steps of reacting systems. However, reactions involving several concerted bond changes─as can be found in many key steps of natural product syntheses─can complicate the search process. To mitigate these complications, we present a mechanism search strategy particularly suited to help expedite exploration of an exemplary family of such complex reactions, cyclizations. We provide a cost-effective strategy for identifying relevant elementary reaction steps by combining graph-based enumeration schemes and machine learning techniques for intermediate filtering. Key to this approach is our use of a neural network potential (NNP), AIMNet2-rxn, for computational evaluation of each candidate reaction pathway. In this article, we evaluate the NNP's ability to estimate activation energies, demonstrate the correct anticipation of stereoselectivity, and recapitulate complex enabling steps in natural product synthesis.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c01161","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Reaction mechanism search tools have demonstrated the ability to provide insights into likely products and rate-limiting steps of reacting systems. However, reactions involving several concerted bond changes─as can be found in many key steps of natural product syntheses─can complicate the search process. To mitigate these complications, we present a mechanism search strategy particularly suited to help expedite exploration of an exemplary family of such complex reactions, cyclizations. We provide a cost-effective strategy for identifying relevant elementary reaction steps by combining graph-based enumeration schemes and machine learning techniques for intermediate filtering. Key to this approach is our use of a neural network potential (NNP), AIMNet2-rxn, for computational evaluation of each candidate reaction pathway. In this article, we evaluate the NNP's ability to estimate activation energies, demonstrate the correct anticipation of stereoselectivity, and recapitulate complex enabling steps in natural product synthesis.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


