{"title":"A novel <i>ANK1</i> frameshift mutation associated with neonatal hereditary spherocytosis: a case report.","authors":"Xin Qing, Jimo Zhu, Xiaoshi Zhu, Yu Zhang, Junchao Deng, Binzhi Tang","doi":"10.3389/fped.2025.1666585","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hereditary spherocytosis (HS) is a genetically inherited hemolytic anemia resulting from erythrocyte membrane defects, predominantly associated with genetic mutations in membrane protein genes such as <i>ANK1</i> and <i>SPTB</i>. The disease exhibits considerable heterogeneity in both clinical manifestations and age of onset, presenting substantial diagnostic challenges for clinicians, particularly in pediatric cases.</p><p><strong>Case presentation: </strong>The patient was a 29-day-old boy who had experienced persistent anemia and a medical history of neonatal hyperbilirubinemia since birth. Upon admission, the infant lacked typical HS manifestations such as splenomegaly, jaundice, and spherocytosis on the peripheral blood smear. Whole-exome sequencing identified a novel frameshift mutation c.3556delG (EX30, NM_000037.4), resulting in an amino acid alteration p.Glu1186Lysfs*3. Subsequent Sanger sequencing-based family segregation analysis confirmed that this mutation originated from the paternal allele. Based on the characteristic clinical manifestations and genetic findings, a definitive diagnosis of HS was established.</p><p><strong>Conclusions: </strong>In neonates presenting with unexplained recurrent anemia, particularly those with a history of neonatal hyperbilirubinemia, HS should be suspected. Due to the atypical manifestations, genetic analysis serves as a pivotal tool in the early diagnosis of HS, and novel genetic mutations may be identified, which can subsequently be added to the genetic database.</p>","PeriodicalId":12637,"journal":{"name":"Frontiers in Pediatrics","volume":"13 ","pages":"1666585"},"PeriodicalIF":2.0000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12488563/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/fped.2025.1666585","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Hereditary spherocytosis (HS) is a genetically inherited hemolytic anemia resulting from erythrocyte membrane defects, predominantly associated with genetic mutations in membrane protein genes such as ANK1 and SPTB. The disease exhibits considerable heterogeneity in both clinical manifestations and age of onset, presenting substantial diagnostic challenges for clinicians, particularly in pediatric cases.
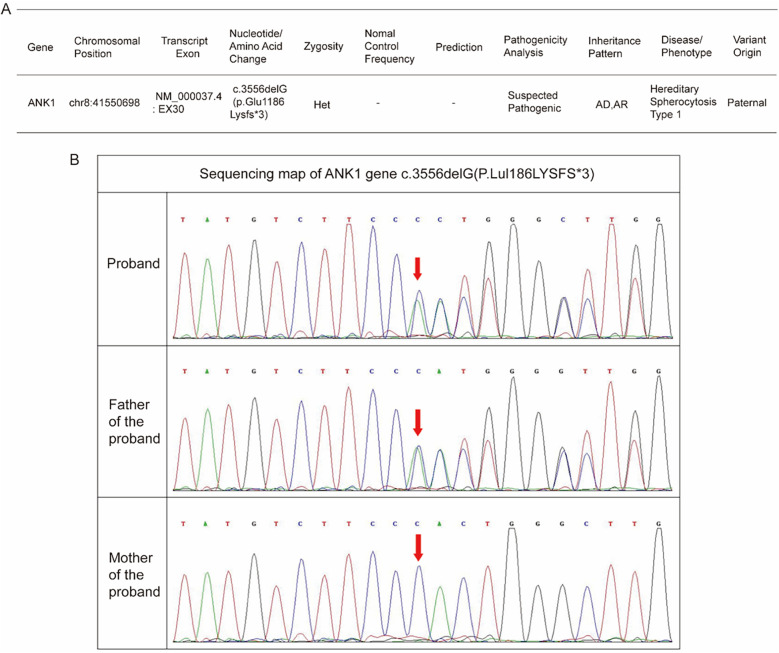
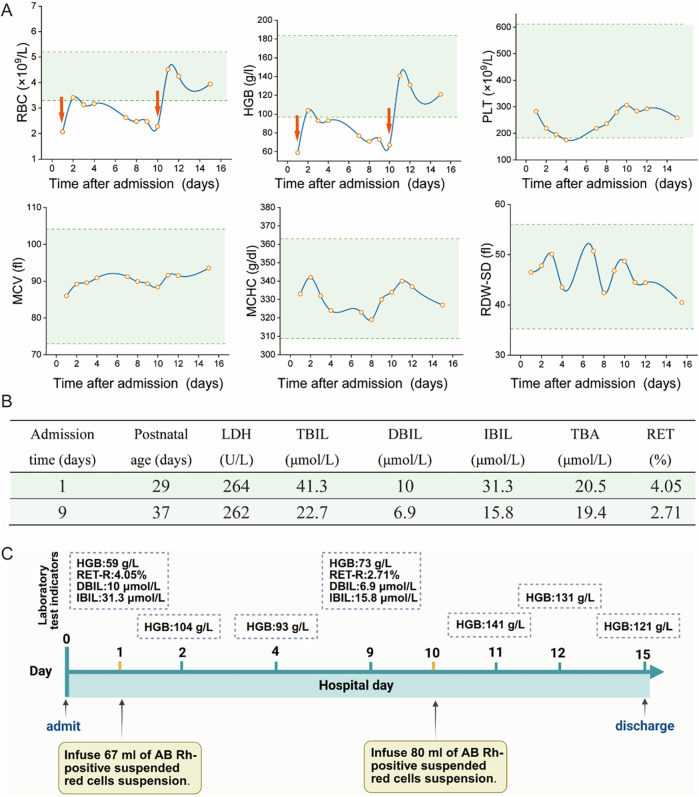
Case presentation: The patient was a 29-day-old boy who had experienced persistent anemia and a medical history of neonatal hyperbilirubinemia since birth. Upon admission, the infant lacked typical HS manifestations such as splenomegaly, jaundice, and spherocytosis on the peripheral blood smear. Whole-exome sequencing identified a novel frameshift mutation c.3556delG (EX30, NM_000037.4), resulting in an amino acid alteration p.Glu1186Lysfs*3. Subsequent Sanger sequencing-based family segregation analysis confirmed that this mutation originated from the paternal allele. Based on the characteristic clinical manifestations and genetic findings, a definitive diagnosis of HS was established.
Conclusions: In neonates presenting with unexplained recurrent anemia, particularly those with a history of neonatal hyperbilirubinemia, HS should be suspected. Due to the atypical manifestations, genetic analysis serves as a pivotal tool in the early diagnosis of HS, and novel genetic mutations may be identified, which can subsequently be added to the genetic database.
期刊介绍:
Frontiers in Pediatrics (Impact Factor 2.33) publishes rigorously peer-reviewed research broadly across the field, from basic to clinical research that meets ongoing challenges in pediatric patient care and child health. Field Chief Editors Arjan Te Pas at Leiden University and Michael L. Moritz at the Children''s Hospital of Pittsburgh are supported by an outstanding Editorial Board of international experts. This multidisciplinary open-access journal is at the forefront of disseminating and communicating scientific knowledge and impactful discoveries to researchers, academics, clinicians and the public worldwide.
Frontiers in Pediatrics also features Research Topics, Frontiers special theme-focused issues managed by Guest Associate Editors, addressing important areas in pediatrics. In this fashion, Frontiers serves as an outlet to publish the broadest aspects of pediatrics in both basic and clinical research, including high-quality reviews, case reports, editorials and commentaries related to all aspects of pediatrics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


