DFT investigation of proton affinity and intramolecular proton–π interactions in N,N-dimethylnaphthylamine derivatives: insights from aromaticity, AIM, and NBO analyses
Hamid Saeidian, Zohreh Mirjafary and Ashkan Ghiasi
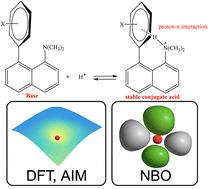
{"title":"DFT investigation of proton affinity and intramolecular proton–π interactions in N,N-dimethylnaphthylamine derivatives: insights from aromaticity, AIM, and NBO analyses","authors":"Hamid Saeidian, Zohreh Mirjafary and Ashkan Ghiasi","doi":"10.1039/D5NJ02742J","DOIUrl":null,"url":null,"abstract":"<p >In this research, the theoretical investigation of intramolecular proton–π (H<small><sup>+</sup></small>⋯π) interactions was conducted on a series of protonated derivatives of <em>N,N</em>-dimethylnaphthalen-1-amine. The primary aim was to examine the characteristics and intensity of these interactions within the conjugate acid structures of the amines. To achieve this, the molecular structures were optimized at the B3LYP/6-311+G(d,p) level of theory in the gas phase and in various solvent environments utilizing the CPCM model. Compounds that include a phenyl ring generally demonstrate proton affinities (PA) exceeding 980 kJ mol<small><sup>−1</sup></small>. The incorporation of strong electron-donating groups, such as amino groups, significantly boosts the basicity, as H<small><sup>+</sup></small>⋯π interactions play a crucial role in enhancing structural stability. The PA value for compound <strong>5</strong>, which possesses a 4-aminophenyl group at the 8-position of the naphthalene core, was determined to be 1007 kJ mol<small><sup>−1</sup></small>, while the PA for the amine with a 3,4-diaminophenyl substituent (compound <strong>6</strong>) rose to 1022 kJ mol<small><sup>−1</sup></small>. Conversely, compounds featuring electron-deficient aromatic rings, such as compound <strong>8</strong> (4-nitrophenyl) and compound <strong>9</strong> (pyridyl), displayed lower basicity and proton affinity compared to the reference compound (compound <strong>4</strong>). In these instances, a significant interaction between the proton and the π-electron system of the phenyl ring is not formed, which explains the reduced stability of the conjugate acid and the diminished proton-accepting ability. Topological analysis based on the atoms in molecules theory (AIM) confirmed the presence of a bond critical point (CP) between the proton and the <em>ipso</em> carbon of the π system. The proton–π distance was found to be less than 2.5 Å, typically in the range of 1.9–2.2 Å. The values of electron density <em>ρ</em>(<em>r</em>) and potential energy <em>V</em>(<em>r</em>) at the BCPs indicated the presence of H<small><sup>+</sup></small>⋯π interactions with varying strengths. Moreover, natural bond orbital analysis revealed significant second-order stabilization energies (up to ∼30 kcal mol<small><sup>−1</sup></small>) associated with charge transfer from π orbitals to the σ*(N–H), especially in <strong>[5 + H]<small><sup>+</sup></small></strong> and <strong>[6 + H]<small><sup>+</sup></small></strong>. These findings were consistent with structural indices such as reduced HOMA and increased SA values in the protonated forms, reflecting a decrease in aromaticity and increased geometric distortion induced by the proton's proximity to the aromatic ring. Overall, this study provides strong and multidimensional theoretical evidence for the presence of H<small><sup>+</sup></small>⋯π interactions in the conjugate acids of aromatic derivatives and comprehensively explores their role in the stability, electron distribution, and structural changes of the designed conjugate acid–base systems.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 39","pages":" 17350-17361"},"PeriodicalIF":2.5000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj02742j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
In this research, the theoretical investigation of intramolecular proton–π (H+⋯π) interactions was conducted on a series of protonated derivatives of N,N-dimethylnaphthalen-1-amine. The primary aim was to examine the characteristics and intensity of these interactions within the conjugate acid structures of the amines. To achieve this, the molecular structures were optimized at the B3LYP/6-311+G(d,p) level of theory in the gas phase and in various solvent environments utilizing the CPCM model. Compounds that include a phenyl ring generally demonstrate proton affinities (PA) exceeding 980 kJ mol−1. The incorporation of strong electron-donating groups, such as amino groups, significantly boosts the basicity, as H+⋯π interactions play a crucial role in enhancing structural stability. The PA value for compound 5, which possesses a 4-aminophenyl group at the 8-position of the naphthalene core, was determined to be 1007 kJ mol−1, while the PA for the amine with a 3,4-diaminophenyl substituent (compound 6) rose to 1022 kJ mol−1. Conversely, compounds featuring electron-deficient aromatic rings, such as compound 8 (4-nitrophenyl) and compound 9 (pyridyl), displayed lower basicity and proton affinity compared to the reference compound (compound 4). In these instances, a significant interaction between the proton and the π-electron system of the phenyl ring is not formed, which explains the reduced stability of the conjugate acid and the diminished proton-accepting ability. Topological analysis based on the atoms in molecules theory (AIM) confirmed the presence of a bond critical point (CP) between the proton and the ipso carbon of the π system. The proton–π distance was found to be less than 2.5 Å, typically in the range of 1.9–2.2 Å. The values of electron density ρ(r) and potential energy V(r) at the BCPs indicated the presence of H+⋯π interactions with varying strengths. Moreover, natural bond orbital analysis revealed significant second-order stabilization energies (up to ∼30 kcal mol−1) associated with charge transfer from π orbitals to the σ*(N–H), especially in [5 + H]+ and [6 + H]+. These findings were consistent with structural indices such as reduced HOMA and increased SA values in the protonated forms, reflecting a decrease in aromaticity and increased geometric distortion induced by the proton's proximity to the aromatic ring. Overall, this study provides strong and multidimensional theoretical evidence for the presence of H+⋯π interactions in the conjugate acids of aromatic derivatives and comprehensively explores their role in the stability, electron distribution, and structural changes of the designed conjugate acid–base systems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


