Single amino acid variation identification in high resolution tandem mass spectrometry data in bottom up proteomics
IF 1.7
3区 化学
Q3 PHYSICS, ATOMIC, MOLECULAR & CHEMICAL
引用次数: 0
Abstract
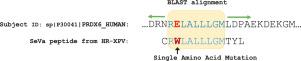
Database-searching based precursor ion identification in tandem mass spectrometry data analysis is limited to the search space. Once a single amino acid variation (SAAV) or a modification is not included to the search space, then its observed spectra will not be annotated correctly. Several methods have been developed to identify and localize post-translational modifications (PTMs); however, few methods have been introduced to identify peptide sequences with amino acid mutations. Here, we present our approach to detect SAAVs, called SeVa (standing for Sequence Variation). SeVa is based on the High-Resolution Exact P-Value (HR-XPV) method (doi:10.1002/pmic.202300145), which builds an exact empirical null distribution by implicitly scoring the spectra against all possible amino acid sequences in high-resolution fragmentation settings. SeVa extracts the amino acid sequence from HR-XPV, which produces the highest score. The SeVa peptides identified are subjected to a homology search against a proteome database containing shuffled decoy protein sequences. This step increases the sensitivity of the results and the decoy identifications can be used to estimate the FDR. We tested SeVa with two experimental datasets related to immunopeptidomics (PXD017407) and cancer (PDC000224), and our method identified 781 and 15,764 peptide sequences with mutations at 1.68% and 0.52% of FDRs.
自底向上蛋白质组学中单氨基酸变异的高分辨率串联质谱鉴定
串联质谱数据分析中基于数据库搜索的前体离子鉴定受限于搜索空间。一旦单个氨基酸变异(SAAV)或修饰不包含在搜索空间中,则其观测光谱将无法正确注释。已经开发了几种方法来识别和定位翻译后修饰(PTMs);然而,很少有方法被引入来鉴定氨基酸突变的肽序列。在这里,我们提出了一种检测saav的方法,称为SeVa(代表序列变异)。SeVa基于高分辨率精确p值(HR-XPV)方法(doi:10.1002/pmic)。202300145),该方法通过对高分辨率片段设置中所有可能的氨基酸序列进行隐式评分,建立了精确的经验零分布。SeVa从HR-XPV中提取的氨基酸序列得分最高。鉴定出的SeVa肽对包含洗牌诱饵蛋白序列的蛋白质组数据库进行同源性搜索。这一步骤提高了结果的灵敏度,并且可以使用诱饵识别来估计FDR。我们使用两个与免疫肽组学(PXD017407)和癌症(PDC000224)相关的实验数据集对SeVa进行了测试,我们的方法鉴定出781和15764个肽序列,突变率分别为1.68%和0.52%。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
3.60
自引率
5.60%
发文量
145
审稿时长
71 days
期刊介绍:
The journal invites papers that advance the field of mass spectrometry by exploring fundamental aspects of ion processes using both the experimental and theoretical approaches, developing new instrumentation and experimental strategies for chemical analysis using mass spectrometry, developing new computational strategies for data interpretation and integration, reporting new applications of mass spectrometry and hyphenated techniques in biology, chemistry, geology, and physics.
Papers, in which standard mass spectrometry techniques are used for analysis will not be considered.
IJMS publishes full-length articles, short communications, reviews, and feature articles including young scientist features.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


