A density functional theory approach for design of fluorine-containing dianionic ionic liquids and investigation of some of their physical and chemical properties
IF 1.9
4区 化学
Q3 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
Abstract
This study used density functional theory (DFT) calculations to investigate the properties of four novel dianionic ionic liquids (DAILs) named as [PMIM]2[TFTP], [PMTA1]2[TFTP], [PMTA2]2[TFTP] and [PMTTA]2[TFTP]. These DAILs consist of a tetrafluoro terephthalate ([TFTP]2-) dianion and various functionalized imidazolium, triazolium and tetrazolium-based cations allowing for an analysis of how cation structure (specifically nitrogen atom number and position) and fluorination affect the DAILs' stability, electronic properties, and electrochemical behavior. The research evaluated energetic, electronic, and thermodynamic parameters, as well as electrostatic potential maps and topological properties, to understand the relationship between DAIL structure and its characteristics. The fluorinated dianion showed a planar preference for interaction with the cation's five-membered rings. Importantly, hydrogen bonding, in conjunction with electrostatic interactions, significantly contributes to the stability of these DAILs and will likely influence their physicochemical properties and suitability for specific applications. The interaction energies for the most stable configurations of the examined DAILs vary between -235.82 and -269.36 kcal mol-1 at M06–2X-GD3/AUG-cc-pVDZ theory level and following a decreasing trend in the order of: [PMIM]2[TFTP] < [PMTA1]2[TFTP] < [PMTA2]2[TFTP] < [PMTTA]2[TFTP]. Additionally, the HOMO–LUMO energy gaps, spanning from 6.49 to 7.38 eV, highlighted notable stability trends in the series. Charge transfer values obtained through NBO analysis were observed between 0.2318 and 0.7307 a.u., reaffirming the contributions of hydrogen bonding.
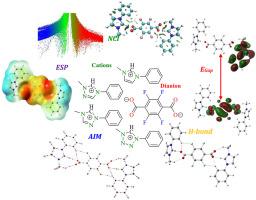
用密度泛函理论方法设计含氟重阴离子液体及其一些理化性质的研究
本研究利用密度泛函理论(DFT)计算研究了四种新型重阴离子液体(DAILs)的性质,分别为[pmmim]2[TFTP]、[PMTA1]2[TFTP]、[PMTA2]2[TFTP]和[PMTTA]2[TFTP]。这些DAILs由四氟对苯二甲酸盐([TFTP]2-)离子和各种功能化的咪唑、三唑和四唑基阳离子组成,可以分析阳离子结构(特别是氮原子序数和位置)和氟化如何影响DAILs的稳定性、电子性能和电化学行为。该研究评估了能量、电子和热力学参数,以及静电势图和拓扑性质,以了解DAIL结构与其特性之间的关系。氟化碘离子表现出与阳离子的五元环相互作用的平面偏好。重要的是,氢键结合静电相互作用,极大地促进了这些dail的稳定性,并可能影响它们的物理化学性质和特定应用的适用性。在m06 - x2 - gd3 /AUG-cc-pVDZ理论水平上,DAILs最稳定构型的相互作用能在-235.82 ~ -269.36 kcal mol-1之间变化,并呈现如下递减趋势:[PMIM]2[TFTP] <; [PMTA1]2[TFTP] < [PMTA2]2[TFTP] < [PMTTA]2[TFTP] < [TFTP] 2[TFTP] < [PMTTA]2[TFTP]] TFTP。此外,HOMO-LUMO的能隙在6.49 ~ 7.38 eV之间,突出了该系列中显著的稳定性趋势。通过NBO分析得到的电荷转移值在0.2318 ~ 0.7307 a.u之间,再次证实了氢键的贡献。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Fluorine Chemistry
化学-无机化学与核化学
CiteScore
3.80
自引率
10.50%
发文量
99
审稿时长
33 days
期刊介绍:
The Journal of Fluorine Chemistry contains reviews, original papers and short communications. The journal covers all aspects of pure and applied research on the chemistry as well as on the applications of fluorine, and of compounds or materials where fluorine exercises significant effects. This can include all chemistry research areas (inorganic, organic, organometallic, macromolecular and physical chemistry) but also includes papers on biological/biochemical related aspects of Fluorine chemistry as well as medicinal, agrochemical and pharmacological research. The Journal of Fluorine Chemistry also publishes environmental and industrial papers dealing with aspects of Fluorine chemistry on energy and material sciences. Preparative and physico-chemical investigations as well as theoretical, structural and mechanistic aspects are covered. The Journal, however, does not accept work of purely routine nature.
For reviews and special issues on particular topics of fluorine chemistry or from selected symposia, please contact the Regional Editors for further details.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


