Carlos M. Bustamante*, , , Franco P. Bonafé, , , Maxim Sukharev, , , Michael Ruggenthaler, , , Abraham Nitzan, , and , Angel Rubio,
{"title":"Molecular Polariton Dynamics in Realistic Cavities","authors":"Carlos M. Bustamante*, , , Franco P. Bonafé, , , Maxim Sukharev, , , Michael Ruggenthaler, , , Abraham Nitzan, , and , Angel Rubio, ","doi":"10.1021/acs.jctc.5c01318","DOIUrl":null,"url":null,"abstract":"<p >The large number of degrees of freedom involved in polaritonic chemistry processes considerably restricts the systems that can be described by any ab initio approach, due to the resulting high computational cost. Semiclassical methods that treat light classically offer a promising route for overcoming these limitations. In this work, we present a new implementation that combines the numerical propagation of Maxwell’s equations to simulate realistic cavities with quantum electron dynamics at the density functional tight-binding (DFTB) theory level. This implementation allows for the simulation of a large number of molecules described at the atomistic level, interacting with cavity modes obtained by numerically solving Maxwell’s equations. By mimicking experimental setups, our approach enables the calculation of transmission spectra, in which we observe the corresponding polaritonic signals. In addition, we have access to local information, revealing complex responses of individual molecules that depend on the number, geometry, position, and orientation of the molecules inside the cavity.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9823–9831"},"PeriodicalIF":5.5000,"publicationDate":"2025-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c01318","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01318","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
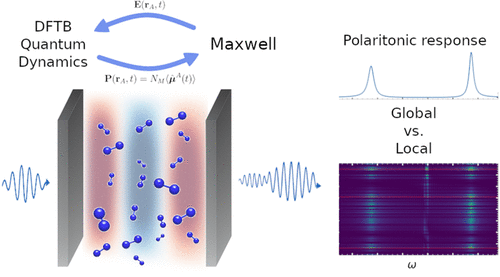
The large number of degrees of freedom involved in polaritonic chemistry processes considerably restricts the systems that can be described by any ab initio approach, due to the resulting high computational cost. Semiclassical methods that treat light classically offer a promising route for overcoming these limitations. In this work, we present a new implementation that combines the numerical propagation of Maxwell’s equations to simulate realistic cavities with quantum electron dynamics at the density functional tight-binding (DFTB) theory level. This implementation allows for the simulation of a large number of molecules described at the atomistic level, interacting with cavity modes obtained by numerically solving Maxwell’s equations. By mimicking experimental setups, our approach enables the calculation of transmission spectra, in which we observe the corresponding polaritonic signals. In addition, we have access to local information, revealing complex responses of individual molecules that depend on the number, geometry, position, and orientation of the molecules inside the cavity.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


