Yeonho Song, , , Minjung Kim, , , Bong June Sung, , and , Jun Soo Kim*,
{"title":"Computational Characterization of DNA Catenanes","authors":"Yeonho Song, , , Minjung Kim, , , Bong June Sung, , and , Jun Soo Kim*, ","doi":"10.1021/acs.jctc.5c01224","DOIUrl":null,"url":null,"abstract":"<p >DNA catenanes are molecular structures composed of two interlocked circular DNA molecules, held together by a mechanical bond─a topological constraint arising from their mutual interlocking. Using all-atom molecular dynamics simulations, we investigated the structural and dynamical properties of DNA catenanes formed by small double-stranded DNA minicircles. In homocatenanes with mild torsional stress (82 bp–82 bp and 92 bp–92 bp), the minicircles largely retain circular conformations, and the mechanical bond exhibits constrained fluctuations in both bond length and twist angle. Rotational diffusion occurs on the microsecond time scale. In the heterocatenane (76 bp–82 bp), elevated torsional stress promotes kink formation in the 76-bp minicircle, leading to a distorted elliptical shape, enhanced DNA–DNA contacts, and anisotropic relaxation characterized by double-exponential decay in both relative translation and twist. Ion distribution analysis shows Na<sup>+</sup> enrichment in the interstitial region between the two DNA minicircles, indicating that counterion condensation also occurs within the interlocked structures. Taken together, these results provide quantitative characterization of relative translation, twisting, and rotation in homocatenanes, while for the heterocatenane the emphasis is placed on qualitative interpretation of anisotropic relaxation. This study highlights how DNA conformation and topological constraints shape the structural and dynamic behavior of DNA catenanes.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9967–9981"},"PeriodicalIF":5.5000,"publicationDate":"2025-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01224","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
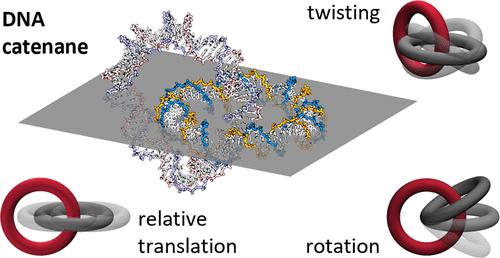
DNA catenanes are molecular structures composed of two interlocked circular DNA molecules, held together by a mechanical bond─a topological constraint arising from their mutual interlocking. Using all-atom molecular dynamics simulations, we investigated the structural and dynamical properties of DNA catenanes formed by small double-stranded DNA minicircles. In homocatenanes with mild torsional stress (82 bp–82 bp and 92 bp–92 bp), the minicircles largely retain circular conformations, and the mechanical bond exhibits constrained fluctuations in both bond length and twist angle. Rotational diffusion occurs on the microsecond time scale. In the heterocatenane (76 bp–82 bp), elevated torsional stress promotes kink formation in the 76-bp minicircle, leading to a distorted elliptical shape, enhanced DNA–DNA contacts, and anisotropic relaxation characterized by double-exponential decay in both relative translation and twist. Ion distribution analysis shows Na+ enrichment in the interstitial region between the two DNA minicircles, indicating that counterion condensation also occurs within the interlocked structures. Taken together, these results provide quantitative characterization of relative translation, twisting, and rotation in homocatenanes, while for the heterocatenane the emphasis is placed on qualitative interpretation of anisotropic relaxation. This study highlights how DNA conformation and topological constraints shape the structural and dynamic behavior of DNA catenanes.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


