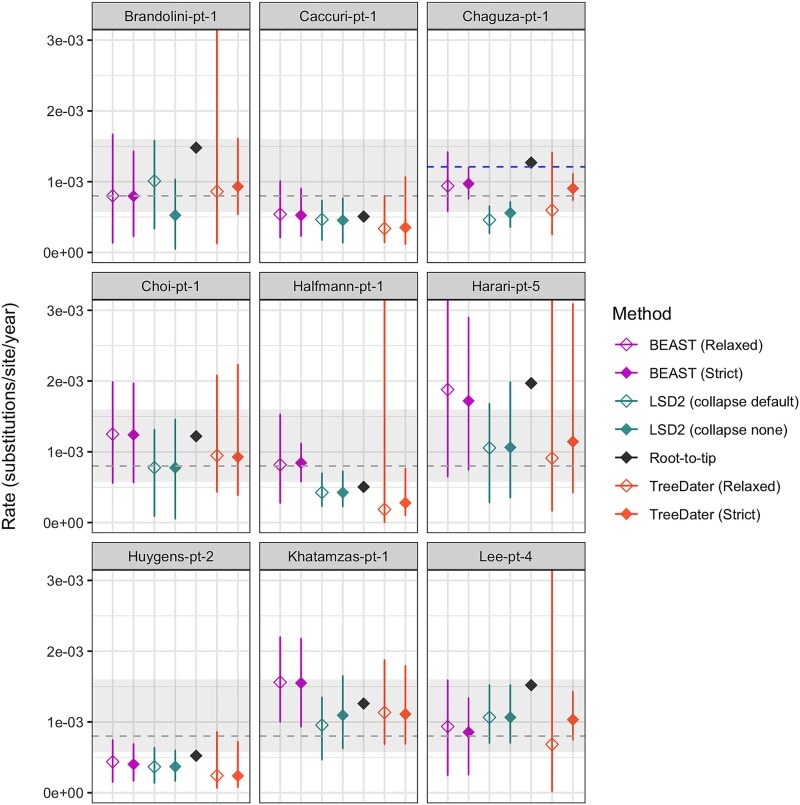
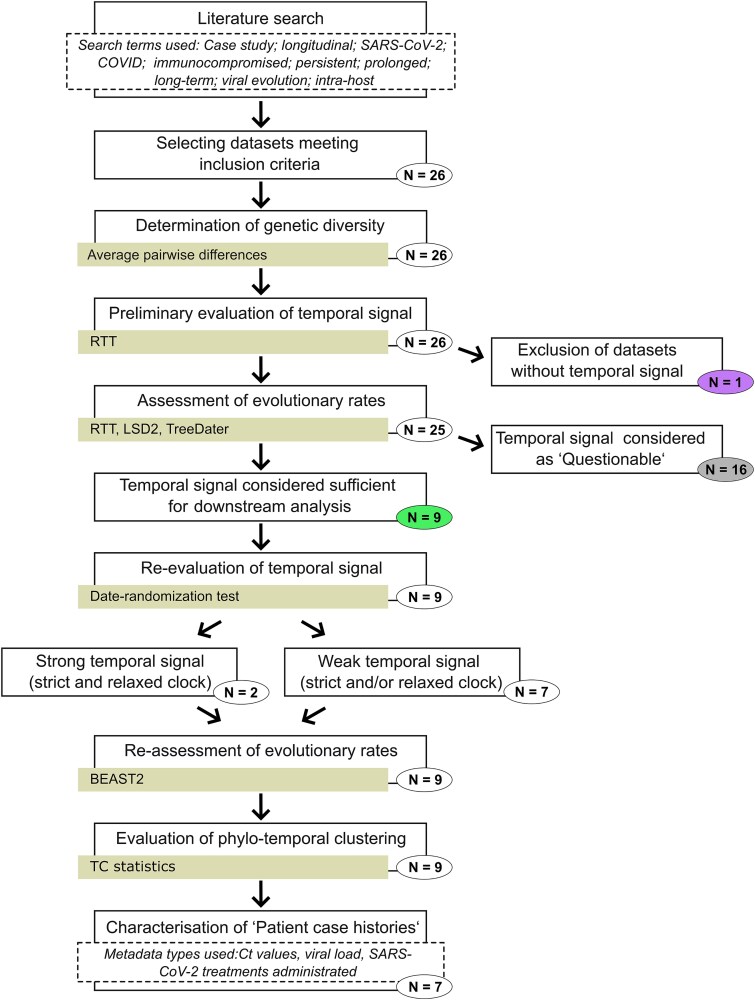
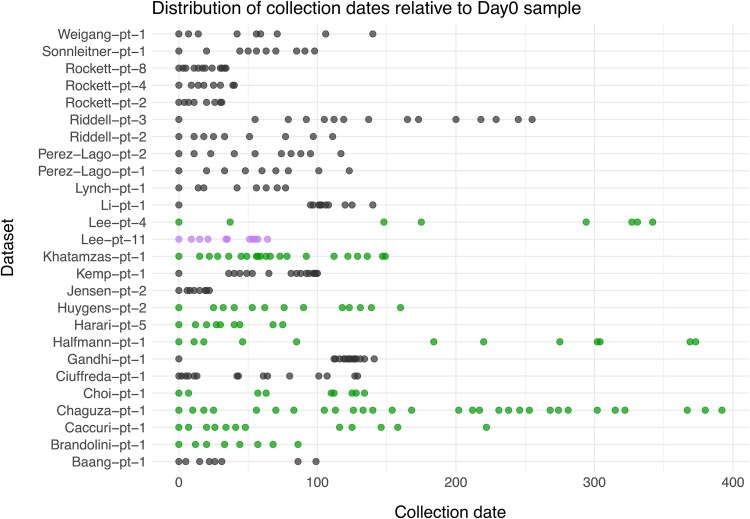
Phylogenetic meta-analysis of persistent SARS-CoV-2 infections in immunocompromised individuals highlights the challenges of robust evolutionary rate estimation caused by low genetic diversity.
Sanni Översti, Emily Gaul, Björn-Erik Ole Jensen, Denise Kühnert
{"title":"Phylogenetic meta-analysis of persistent SARS-CoV-2 infections in immunocompromised individuals highlights the challenges of robust evolutionary rate estimation caused by low genetic diversity.","authors":"Sanni Översti, Emily Gaul, Björn-Erik Ole Jensen, Denise Kühnert","doi":"10.1093/ve/veaf065","DOIUrl":null,"url":null,"abstract":"<p><p>Time-stamped genomic sequences from rapidly evolving pathogens can be used to estimate the rates of evolution through molecular tip-dating. The validity of this approach, however, depends on whether detectable levels of genetic variation have accumulated over the given sampling interval, generating a temporal signal. Moreover, molecular dating methods have demonstrated varying degrees of systematic biases under different biologically realistic scenarios, such as the presence of phylo-temporal clustering. Persistent SARS-CoV-2 infections in immunocompromised individuals have been linked to accelerated intrahost molecular rates compared to those of global lineages, facilitating the emergence of novel viral lineages. Yet, studies reporting elevated rates lack assessment of data properties, such as evaluation of temporal signal and comparison of multiple methods of inference, both crucial for robust rate estimation. In this study, we applied a range of molecular dating approaches to reassess the rate of SARS-CoV-2 intrahost evolution in immunocompromised individuals using publicly available datasets. Our findings suggest that even during long-term infections, the limited number of genetic changes accumulating may pose a challenge for robust inference of within-host evolutionary rates, particularly when relying on consensus sequences and when datasets are small or unevenly sampled. Moreover, our results highlight that when certain methodological limitations are overlooked, evolutionary rates can be significantly overestimated. In general, our findings demonstrate that estimating within-host evolutionary rates is a challenging question necessitating thorough assessment of data quality, careful selection of appropriate methods, and cautious interpretation of the resulting estimates. Whereas our phylogenetic analyses of viral consensus sequences provide no evidence of elevated evolutionary rates across the complete genome during chronic SARS-CoV-2 infection, prolonged viral shedding may nevertheless promote the emergence of new viral variants in immunocompromised individuals.</p>","PeriodicalId":56026,"journal":{"name":"Virus Evolution","volume":"11 1","pages":"veaf065"},"PeriodicalIF":4.0000,"publicationDate":"2025-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12477587/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Evolution","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/ve/veaf065","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Time-stamped genomic sequences from rapidly evolving pathogens can be used to estimate the rates of evolution through molecular tip-dating. The validity of this approach, however, depends on whether detectable levels of genetic variation have accumulated over the given sampling interval, generating a temporal signal. Moreover, molecular dating methods have demonstrated varying degrees of systematic biases under different biologically realistic scenarios, such as the presence of phylo-temporal clustering. Persistent SARS-CoV-2 infections in immunocompromised individuals have been linked to accelerated intrahost molecular rates compared to those of global lineages, facilitating the emergence of novel viral lineages. Yet, studies reporting elevated rates lack assessment of data properties, such as evaluation of temporal signal and comparison of multiple methods of inference, both crucial for robust rate estimation. In this study, we applied a range of molecular dating approaches to reassess the rate of SARS-CoV-2 intrahost evolution in immunocompromised individuals using publicly available datasets. Our findings suggest that even during long-term infections, the limited number of genetic changes accumulating may pose a challenge for robust inference of within-host evolutionary rates, particularly when relying on consensus sequences and when datasets are small or unevenly sampled. Moreover, our results highlight that when certain methodological limitations are overlooked, evolutionary rates can be significantly overestimated. In general, our findings demonstrate that estimating within-host evolutionary rates is a challenging question necessitating thorough assessment of data quality, careful selection of appropriate methods, and cautious interpretation of the resulting estimates. Whereas our phylogenetic analyses of viral consensus sequences provide no evidence of elevated evolutionary rates across the complete genome during chronic SARS-CoV-2 infection, prolonged viral shedding may nevertheless promote the emergence of new viral variants in immunocompromised individuals.
期刊介绍:
Virus Evolution is a new Open Access journal focusing on the long-term evolution of viruses, viruses as a model system for studying evolutionary processes, viral molecular epidemiology and environmental virology.
The aim of the journal is to provide a forum for original research papers, reviews, commentaries and a venue for in-depth discussion on the topics relevant to virus evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


