Dielectric Anisotropy Mitigated by Hydrogen Bonding Governs the Driving Force of Charge Transport within Quinone Pairs in Photosynthetic Reaction Centers
Muhammet Erkan Köse, , , Roshan Khatri, , and , Barry D. Dunietz*,
{"title":"Dielectric Anisotropy Mitigated by Hydrogen Bonding Governs the Driving Force of Charge Transport within Quinone Pairs in Photosynthetic Reaction Centers","authors":"Muhammet Erkan Köse, , , Roshan Khatri, , and , Barry D. Dunietz*, ","doi":"10.1021/acs.jpca.5c04134","DOIUrl":null,"url":null,"abstract":"<p >In a bacterial photosynthetic reaction center (bRC) and photosystem II (PSII), the charge transport (CT) is between the primary semiquinone (<i>Q</i><sub>A</sub>) and the secondary quinone (<i>Q</i><sub>B</sub>). The doubly reduced form of <i>Q</i><sub>B</sub> is converted upon protonation to a labile quinol that leaves the binding site. Despite the chemical and structural similarities of the quinone pairs in the two reaction centers (RCs), the free energy change affecting the quinone CT differs significantly due to asymmetries in their electrostatic environment, molecular conformation, and nonbonding interactions with nearby cofactors and protein residues. This study aims to quantify the energetic contributions of these factors to the driving force for the CT between the quinones, resolve the key nonbonded interactions with nearby units, and explain differences in the energetics between the RCs. Toward this goal, we use a screened range-separated hybrid functional, which correctly accounts for polarizable environment. In particular, dielectric anisotropy is found to be the largest contributor to the free energy change of CT in both RCs. Hydrogen-bonding interactions around quinones are identified, and their stabilization is evaluated by accounting for the effective dielectric constant of the surrounding environment. By resolving the free energy contributions, we identify the particular hydrogen-bonding networks affecting the quinones and which determine the reduction midpoint potential differences. In both RCs, the quinones appear to form hydrogen bonds with three protein residue units, of which histidines bear the strongest bond. In considering the forward transport process, we find for both RCs that the anionic <i>Q</i><sub>A</sub> is hydrogen-bonded with all nearby protein residues, while the labile <i>Q</i><sub>B</sub> in the neutral states appears to exclude the nearby serine residues from such interactions. The calculated free energy change for CT agrees remarkably well with experimental findings.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 40","pages":"9163–9175"},"PeriodicalIF":2.8000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c04134","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
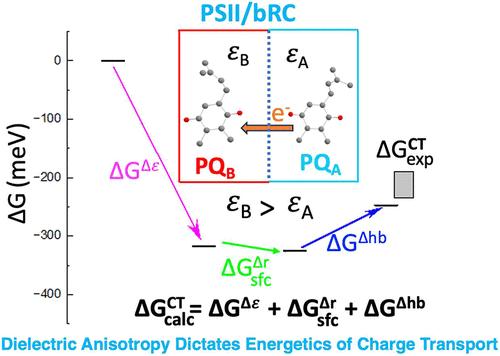
In a bacterial photosynthetic reaction center (bRC) and photosystem II (PSII), the charge transport (CT) is between the primary semiquinone (QA) and the secondary quinone (QB). The doubly reduced form of QB is converted upon protonation to a labile quinol that leaves the binding site. Despite the chemical and structural similarities of the quinone pairs in the two reaction centers (RCs), the free energy change affecting the quinone CT differs significantly due to asymmetries in their electrostatic environment, molecular conformation, and nonbonding interactions with nearby cofactors and protein residues. This study aims to quantify the energetic contributions of these factors to the driving force for the CT between the quinones, resolve the key nonbonded interactions with nearby units, and explain differences in the energetics between the RCs. Toward this goal, we use a screened range-separated hybrid functional, which correctly accounts for polarizable environment. In particular, dielectric anisotropy is found to be the largest contributor to the free energy change of CT in both RCs. Hydrogen-bonding interactions around quinones are identified, and their stabilization is evaluated by accounting for the effective dielectric constant of the surrounding environment. By resolving the free energy contributions, we identify the particular hydrogen-bonding networks affecting the quinones and which determine the reduction midpoint potential differences. In both RCs, the quinones appear to form hydrogen bonds with three protein residue units, of which histidines bear the strongest bond. In considering the forward transport process, we find for both RCs that the anionic QA is hydrogen-bonded with all nearby protein residues, while the labile QB in the neutral states appears to exclude the nearby serine residues from such interactions. The calculated free energy change for CT agrees remarkably well with experimental findings.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


