InterPLM: discovering interpretable features in protein language models via sparse autoencoders
IF 32.1
1区 生物学
Q1 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Despite their success in protein modeling and design, the internal mechanisms of protein language models (PLMs) are poorly understood. Here we present a systematic framework to extract and analyze interpretable features from PLMs using sparse autoencoders. Training sparse autoencoders on ESM-2 embeddings, we identify thousands of interpretable features highlighting biological concepts including binding sites, structural motifs and functional domains. Individual neurons show considerably less conceptual alignment, suggesting PLMs store concepts in superposition. This superposition persists across model scales and larger PLMs capture more interpretable concepts. Beyond known annotations, ESM-2 learns coherent patterns across evolutionarily distinct protein families. To systematically analyze these numerous features, we developed an automated interpretation approach using large language models for feature description and validation. As practical applications, these features can accurately identify missing database annotations and enable targeted steering of sequence generation. Our results show PLM representations can be decomposed into interpretable components, demonstrating the feasibility and utility of mechanistically interpreting these models. InterPLM is a computational framework to extract and analyze interpretable features from protein language models using sparse autoencoders. By training sparse autoencoders on ESM-2 embeddings, this study identifies thousands of interpretable biological features learned by the different layers of the ESM-2 model.
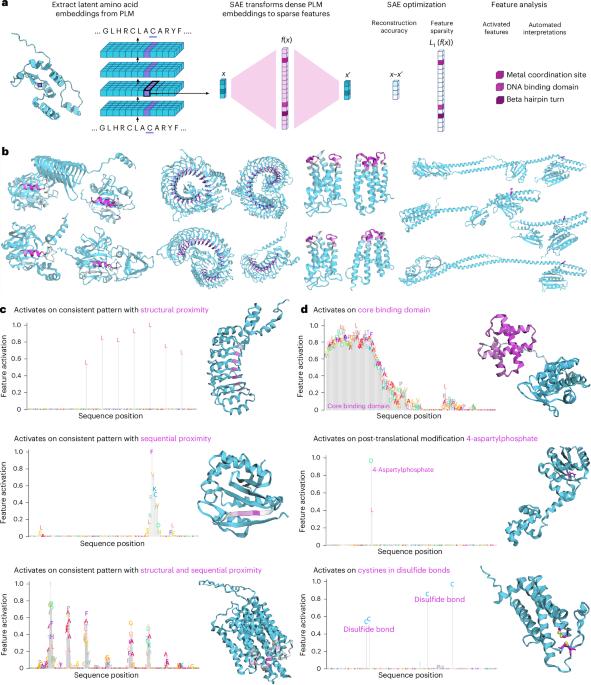
InterPLM:通过稀疏自编码器发现蛋白质语言模型中的可解释特征。
尽管在蛋白质建模和设计方面取得了成功,但人们对蛋白质语言模型(PLMs)的内部机制知之甚少。在这里,我们提出了一个系统的框架,以提取和分析可解释的特征,从plm使用稀疏自编码器。在ESM-2嵌入上训练稀疏自编码器,我们识别出数千个突出生物学概念的可解释特征,包括结合位点、结构基序和功能域。单个神经元表现出的概念一致性要少得多,这表明plm以叠加的方式存储概念。这种叠加在模型尺度上持续存在,更大的plm捕获更多可解释的概念。除了已知的注释,ESM-2在进化上不同的蛋白质家族中学习连贯的模式。为了系统地分析这些众多的特征,我们开发了一种使用大型语言模型进行特征描述和验证的自动解释方法。作为实际应用,这些特性可以准确地识别缺失的数据库注释,并实现有针对性的序列生成。我们的研究结果表明,PLM表示可以分解为可解释的组件,证明了机械解释这些模型的可行性和实用性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Methods
生物-生化研究方法
CiteScore
58.70
自引率
1.70%
发文量
326
审稿时长
1 months
期刊介绍:
Nature Methods is a monthly journal that focuses on publishing innovative methods and substantial enhancements to fundamental life sciences research techniques. Geared towards a diverse, interdisciplinary readership of researchers in academia and industry engaged in laboratory work, the journal offers new tools for research and emphasizes the immediate practical significance of the featured work. It publishes primary research papers and reviews recent technical and methodological advancements, with a particular interest in primary methods papers relevant to the biological and biomedical sciences. This includes methods rooted in chemistry with practical applications for studying biological problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


