Modeling graphene oxide nanoribbons: A first-principles study
IF 4.9
3区 材料科学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
In this study, we modeled a graphene oxide nanoribbon (GONR) using first-principles calculations based on Density Functional Theory. The construction of the GONR involved examining the interactions of the oxygen functional groups (OFGs) -O-, -OH, -CO, and -COOH with the graphene nanoribbon (GNR), as well as their interactions with one another through direct contact or mediated by the GNR. To understand the changes experienced by the system when adsorbing different OFGs, we analyzed structural modifications and electronic properties by exploring the band structure, HOMO and LUMO frontier orbitals, and the resulting magnetization. We found that the adsorption of these OFGs occurs preferentially at the edges and near other groups. The concentration of OFGs at each edge affects the electronic properties, which are reflected in the active sites defined by the isosurfaces of the frontier orbitals. The creation of a monovacancy in the final configuration of the GONR leads to a shift in the electronic bands around the Fermi level. These changes enhance the density and distribution of active sites, which are primarily dominated by the vacancy but also present throughout the nanoribbon. These results suggest its potential application in studying - interactions when adsorbing organic molecules.
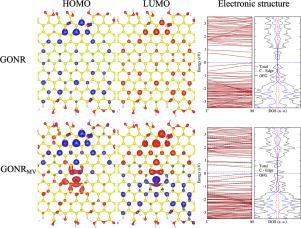
氧化石墨烯纳米带建模:第一性原理研究
在这项研究中,我们利用基于密度泛函理论的第一性原理计算模拟了氧化石墨烯纳米带(GONR)。GONR的构建包括检测氧官能团(ofg) - o -、- oh、- co和- cooh与石墨烯纳米带(GNR)的相互作用,以及它们通过直接接触或在GNR的介导下相互作用。为了了解系统在吸附不同OFGs时所经历的变化,我们通过探索能带结构、HOMO和LUMO前沿轨道以及由此产生的磁化强度来分析结构修饰和电子性能。我们发现这些OFGs的吸附优先发生在边缘和其他基团附近。各边缘ofg的浓度影响电子性质,这反映在前沿轨道等值面定义的活性位点上。在GONR的最终构型中产生的单空位导致费米能级周围电子带的移位。这些变化增强了活性位点的密度和分布,这些活性位点主要以空位为主,但也存在于整个纳米带中。这些结果表明它在研究吸附有机分子时π-π相互作用方面具有潜在的应用前景。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
7.80
自引率
2.50%
发文量
605
审稿时长
40 days
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


