Rashid Mir, Mohammad Fahad Ullah, Imadeldin Elfaki, Mohammad A Alanazi, Naseh A Algehainy, Faisal H Altemani, Mamdoh S Moawadh, Faris J Tayeb, Badr A Alsayed, Mohammad Muzaffar Mir, Jaber Alfaifi, Syed Khalid Mustafa, Jameel Barnawi, Salma Saleh Alrdahe
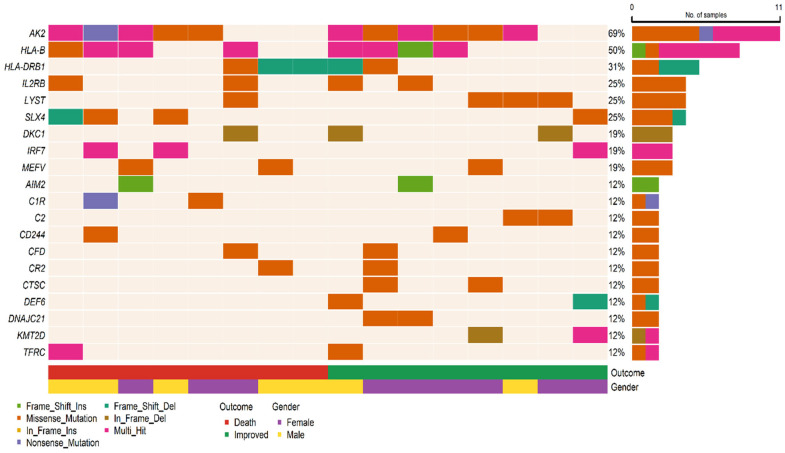
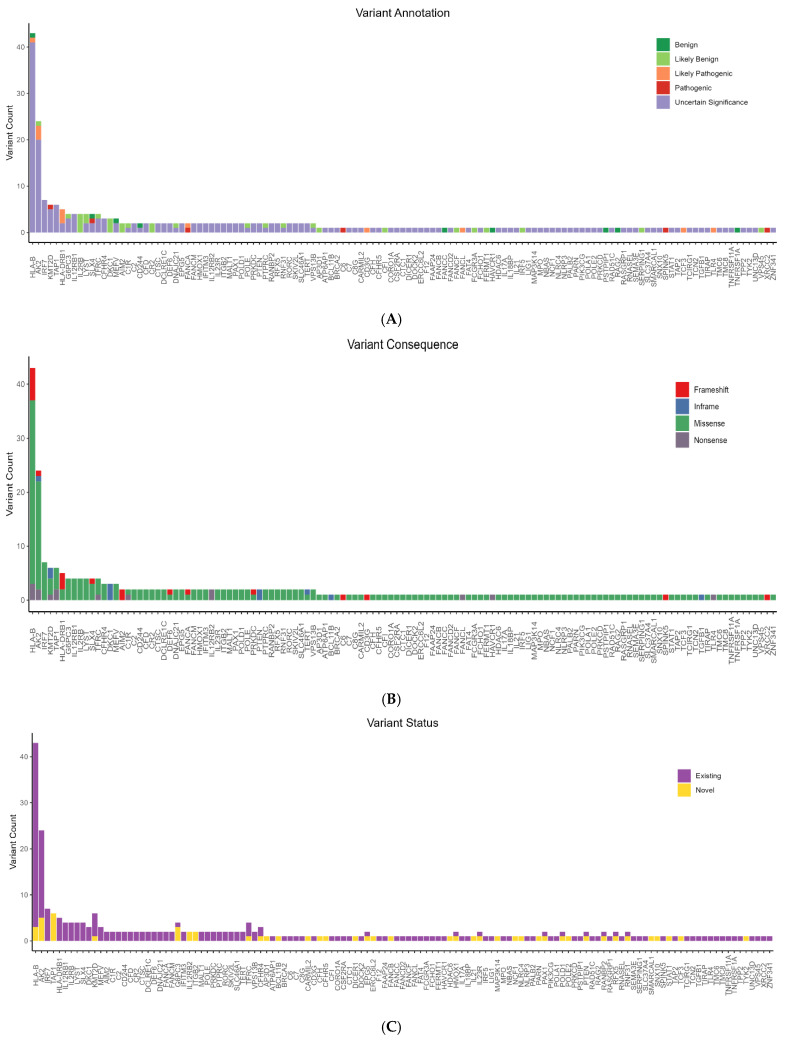
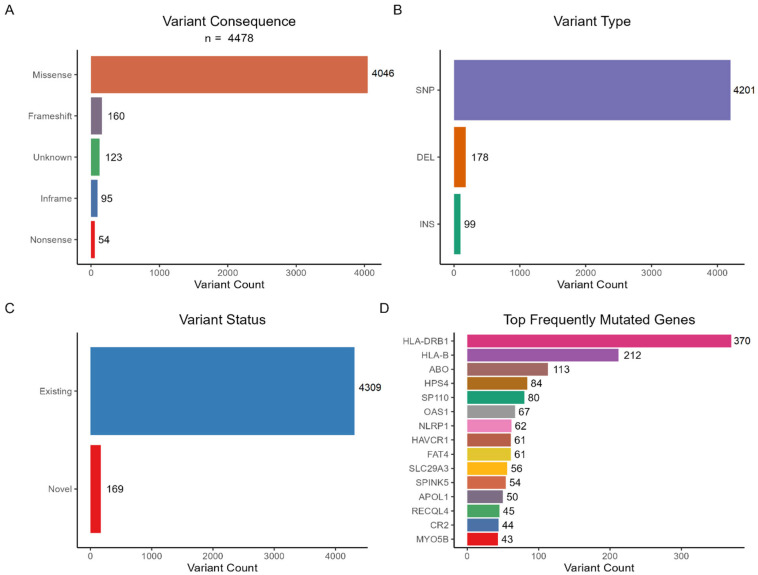
{"title":"Whole-Exome Sequencing Reveals Rare Genetic Variants in Saudi COVID-19 Patients with Extreme Phenotypes.","authors":"Rashid Mir, Mohammad Fahad Ullah, Imadeldin Elfaki, Mohammad A Alanazi, Naseh A Algehainy, Faisal H Altemani, Mamdoh S Moawadh, Faris J Tayeb, Badr A Alsayed, Mohammad Muzaffar Mir, Jaber Alfaifi, Syed Khalid Mustafa, Jameel Barnawi, Salma Saleh Alrdahe","doi":"10.3390/v17091198","DOIUrl":null,"url":null,"abstract":"<p><p>The global impact of COVID-19 was staggering, with millions of cases and related mortality reported worldwide. Genetic variations play a significant role in determining an individual's susceptibility to SARS-CoV-2 infection and progress to severe disease. This pilot study provides an experimental approach using WES to identify certain rare and novel genetic variants that might affect an individual's susceptibility to the risk of SARS-CoV-2 infection, offering an initial exploration of these genetic variants. In the study cohort with 16 patients, the mortality rate was higher in male patients due to severe disease. There was a substantial burden of comorbidity, including hypertension, ischemic heart disease, and T2DM, conditions which independently increase the risk of adverse outcomes in COVID-19 patients. A total of 4478 variants were identified, distributed across 322 genes within the cohort. The majority of these variants were missense substitutions along with frameshift variants, inframe insertions/deletions (indels), and nonsense variants. The variants were further categorized by types to include single-nucleotide polymorphisms (SNPs), deletions (DEL), and insertions (INS). The gene with the highest number of variants was <i>HLA-DRB1</i>, followed by <i>HLA-B</i>, <i>ABO</i>, <i>HPS4</i>, and <i>SP110</i> displaying both common polymorphisms and rare variants. Moreover, the <i>HLA-B</i> gene exhibited the highest number of rare candidate variants, followed by <i>AK2</i>, <i>IRF7</i>, <i>KMT2D</i>, <i>TAP1</i>, and <i>HLA-DRB1</i>. Several genes harbored multiple novel variants, including <i>TAP1</i>, <i>AK2</i>, <i>G6PC3</i>, <i>HLA-B</i>, <i>IL12RB2</i>, and <i>ITGB2</i>. The frequencies of the identified variants were found to be either zero or extremely low (below 1% threshold) in the Middle Eastern or in the overall combined population, suggesting that these are indeed rare and do not represent common indigenous polymorphisms. Functional enrichment analysis of the constructed protein-protein interaction network in our preliminary findings revealed that the identified genes are primarily enriched in pathways associated with immune deficiency and DNA repair. This initial exploration of genetic variants in COVID-19 susceptibility provides a foundation for future large-scale studies.</p>","PeriodicalId":49328,"journal":{"name":"Viruses-Basel","volume":"17 9","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2025-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12474163/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Viruses-Basel","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3390/v17091198","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
The global impact of COVID-19 was staggering, with millions of cases and related mortality reported worldwide. Genetic variations play a significant role in determining an individual's susceptibility to SARS-CoV-2 infection and progress to severe disease. This pilot study provides an experimental approach using WES to identify certain rare and novel genetic variants that might affect an individual's susceptibility to the risk of SARS-CoV-2 infection, offering an initial exploration of these genetic variants. In the study cohort with 16 patients, the mortality rate was higher in male patients due to severe disease. There was a substantial burden of comorbidity, including hypertension, ischemic heart disease, and T2DM, conditions which independently increase the risk of adverse outcomes in COVID-19 patients. A total of 4478 variants were identified, distributed across 322 genes within the cohort. The majority of these variants were missense substitutions along with frameshift variants, inframe insertions/deletions (indels), and nonsense variants. The variants were further categorized by types to include single-nucleotide polymorphisms (SNPs), deletions (DEL), and insertions (INS). The gene with the highest number of variants was HLA-DRB1, followed by HLA-B, ABO, HPS4, and SP110 displaying both common polymorphisms and rare variants. Moreover, the HLA-B gene exhibited the highest number of rare candidate variants, followed by AK2, IRF7, KMT2D, TAP1, and HLA-DRB1. Several genes harbored multiple novel variants, including TAP1, AK2, G6PC3, HLA-B, IL12RB2, and ITGB2. The frequencies of the identified variants were found to be either zero or extremely low (below 1% threshold) in the Middle Eastern or in the overall combined population, suggesting that these are indeed rare and do not represent common indigenous polymorphisms. Functional enrichment analysis of the constructed protein-protein interaction network in our preliminary findings revealed that the identified genes are primarily enriched in pathways associated with immune deficiency and DNA repair. This initial exploration of genetic variants in COVID-19 susceptibility provides a foundation for future large-scale studies.
期刊介绍:
Viruses (ISSN 1999-4915) is an open access journal which provides an advanced forum for studies of viruses. It publishes reviews, regular research papers, communications, conference reports and short notes. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. There is no restriction on the length of the papers. The full experimental details must be provided so that the results can be reproduced. We also encourage the publication of timely reviews and commentaries on topics of interest to the virology community and feature highlights from the virology literature in the ''News and Views'' section. Electronic files or software regarding the full details of the calculation and experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


