Bushran N Iqbal, Sibra R M Shihab, Tao Zhang, Aadhil Ahamed, Shiyamalee Arunasalam, Samanthika Jagoda, Leo L M Poon, Malik Peiris, Faseeha Noordeen
{"title":"Molecular Epidemiology of SARS-CoV-2 Detected from Different Areas of the Kandy District of Sri Lanka from November 2020-March 2022.","authors":"Bushran N Iqbal, Sibra R M Shihab, Tao Zhang, Aadhil Ahamed, Shiyamalee Arunasalam, Samanthika Jagoda, Leo L M Poon, Malik Peiris, Faseeha Noordeen","doi":"10.3390/v17091189","DOIUrl":null,"url":null,"abstract":"<p><p>A comprehensive analysis of the molecular epidemiology of SARS-CoV-2 in the Kandy District of Sri Lanka from November 2020 to March 2022 was conducted to address the limited genomic surveillance data available across the country. The study investigated the circulating SARS-CoV-2 lineages, their temporal dynamics, and the associated mutational profiles in the study area. A total of 280 SARS-CoV-2-positive samples were selected, and 252 complete genomes were successfully sequenced using Oxford Nanopore Technology. Lineage classification was performed using the EPI2ME tool, while phylogenetic relationships were inferred through maximum likelihood and time-scaled phylogenetic trees using IQ-TREE2 and BEAST, respectively. Amino acid substitutions were analyzed to understand lineage-specific mutation patterns. Fifteen SARS-CoV-2 lineages were identified, and of those B.1.411 (36%) was the most prevalent, followed by Q.8 (21%), AY.28 (9.5%), and the Delta and Omicron variants. The lineage distribution showed a temporal shift from B.1.411 to Alpha, Delta, and finally the Omicron, mirroring the global trends. Time to the most recent common ancestor analyses provided estimates for the introduction of major variants, while mutation analysis revealed the widespread occurrence of D614G in the spike protein and lineage-specific mutations across structural, non-structural, and accessory proteins.Detection of the Epsilon variant (absent in other national-level studies) in November 2020, highlighted the regional heterogeneity viral spread. This study emphasizes the importance of localized genomic surveillance to capture the true diversity and evolution of SARS-CoV-2, to facilitate containment strategies in resource-limited settings.</p>","PeriodicalId":49328,"journal":{"name":"Viruses-Basel","volume":"17 9","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2025-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12474409/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Viruses-Basel","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3390/v17091189","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
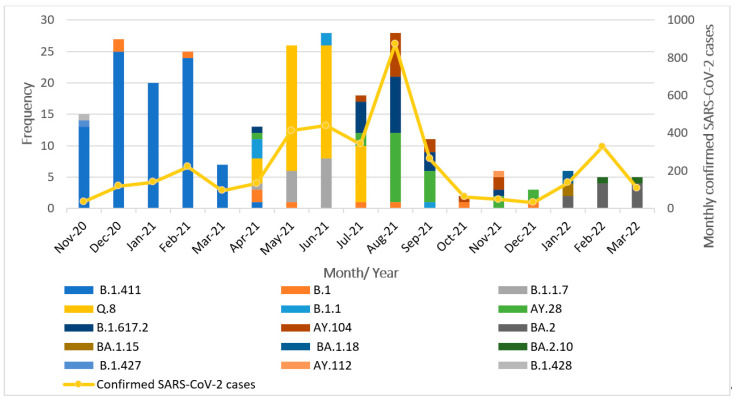
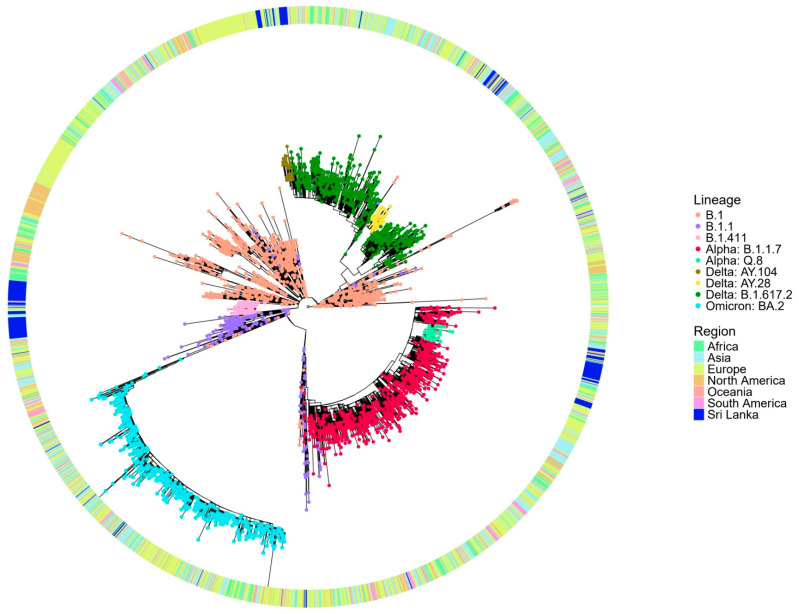
A comprehensive analysis of the molecular epidemiology of SARS-CoV-2 in the Kandy District of Sri Lanka from November 2020 to March 2022 was conducted to address the limited genomic surveillance data available across the country. The study investigated the circulating SARS-CoV-2 lineages, their temporal dynamics, and the associated mutational profiles in the study area. A total of 280 SARS-CoV-2-positive samples were selected, and 252 complete genomes were successfully sequenced using Oxford Nanopore Technology. Lineage classification was performed using the EPI2ME tool, while phylogenetic relationships were inferred through maximum likelihood and time-scaled phylogenetic trees using IQ-TREE2 and BEAST, respectively. Amino acid substitutions were analyzed to understand lineage-specific mutation patterns. Fifteen SARS-CoV-2 lineages were identified, and of those B.1.411 (36%) was the most prevalent, followed by Q.8 (21%), AY.28 (9.5%), and the Delta and Omicron variants. The lineage distribution showed a temporal shift from B.1.411 to Alpha, Delta, and finally the Omicron, mirroring the global trends. Time to the most recent common ancestor analyses provided estimates for the introduction of major variants, while mutation analysis revealed the widespread occurrence of D614G in the spike protein and lineage-specific mutations across structural, non-structural, and accessory proteins.Detection of the Epsilon variant (absent in other national-level studies) in November 2020, highlighted the regional heterogeneity viral spread. This study emphasizes the importance of localized genomic surveillance to capture the true diversity and evolution of SARS-CoV-2, to facilitate containment strategies in resource-limited settings.
期刊介绍:
Viruses (ISSN 1999-4915) is an open access journal which provides an advanced forum for studies of viruses. It publishes reviews, regular research papers, communications, conference reports and short notes. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. There is no restriction on the length of the papers. The full experimental details must be provided so that the results can be reproduced. We also encourage the publication of timely reviews and commentaries on topics of interest to the virology community and feature highlights from the virology literature in the ''News and Views'' section. Electronic files or software regarding the full details of the calculation and experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


