Xinchun Wu, , , Xuezhi Bian, , , Jonathan Rawlinson, , , Robert G. Littlejohn, , and , Joseph E. Subotnik*,
{"title":"Recovering Exact Vibrational Energies within a Phase Space Electronic Structure Framework","authors":"Xinchun Wu, , , Xuezhi Bian, , , Jonathan Rawlinson, , , Robert G. Littlejohn, , and , Joseph E. Subotnik*, ","doi":"10.1021/acs.jctc.5c00956","DOIUrl":null,"url":null,"abstract":"<p >In recent years, there has been a push to go beyond the Born–Oppenheimer theory and build electronic states from a phase space perspective, i.e., parametrize electronic states by both nuclear position (<b><i>R</i></b>) and nuclear momentum (<b><i>P</i></b>). Previous empirical studies have demonstrated that such approaches can yield improved single-surface observables, including vibrational energies, electronic momenta, and vibrational circular dichroism spectra. That being said, unlike the case of the BO theory, there is no unique phase space electronic Hamiltonian nor any theory for using phase space eigenvectors (as opposed to BO eigenvectors) to recover exact quantum vibrational eigenvalues. As such, one might consider such phase space approaches <i>ad hoc</i>. To that end, here we show how to formally extract exact quantum energies from a coupled nuclear-electronic Hamiltonian using perturbation theory on top of a phase space electronic framework. Thus, while we cannot isolate an “optimal” phase space electronic Hamiltonian, this work does justify a phase space electronic structure approach by offering a rigorous framework for correcting the zeroth-order phase space electronic states.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9470–9482"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00956","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
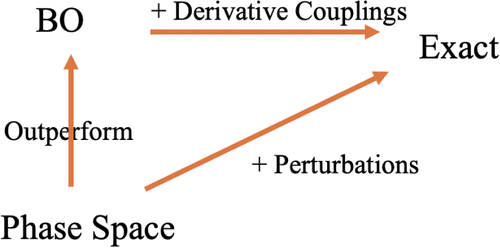
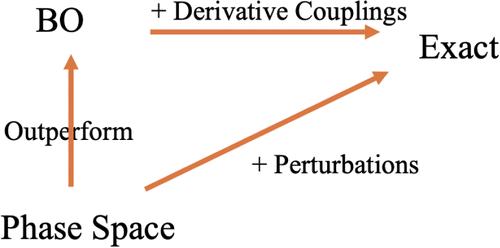
In recent years, there has been a push to go beyond the Born–Oppenheimer theory and build electronic states from a phase space perspective, i.e., parametrize electronic states by both nuclear position (R) and nuclear momentum (P). Previous empirical studies have demonstrated that such approaches can yield improved single-surface observables, including vibrational energies, electronic momenta, and vibrational circular dichroism spectra. That being said, unlike the case of the BO theory, there is no unique phase space electronic Hamiltonian nor any theory for using phase space eigenvectors (as opposed to BO eigenvectors) to recover exact quantum vibrational eigenvalues. As such, one might consider such phase space approaches ad hoc. To that end, here we show how to formally extract exact quantum energies from a coupled nuclear-electronic Hamiltonian using perturbation theory on top of a phase space electronic framework. Thus, while we cannot isolate an “optimal” phase space electronic Hamiltonian, this work does justify a phase space electronic structure approach by offering a rigorous framework for correcting the zeroth-order phase space electronic states.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


