{"title":"Coupled-Cluster in Density Functional Theory Embedding Applied to Static Polarizabilities in Aqueous Environments","authors":"Anthuan Ferino-Pérez*, and , Thomas-C. Jagau, ","doi":"10.1021/acs.jctc.5c00767","DOIUrl":null,"url":null,"abstract":"<p >We present a study of static polarizabilities of organic molecules in aqueous environments using projection-based coupled-cluster in density functional theory quantum embedding. We propose two methods for the computation of supermolecular polarizabilities: an iterative embedding approach and a finite-field approach. The performance of these methods is tested against regular coupled-cluster singles and doubles (CCSD) theory. The static polarizability tensor of the investigated organic molecules varies only slightly with the inclusion of water molecules. The iterative CCSD-in-DFT approach produces isotropic polarizabilities of CCSD quality with mean relative errors smaller than 1.0% at a reduced computational cost, while the anisotropic polarizabilities are not as well described. The choice of the exchange correlation functional for the treatment of the environment has little impact on the quality of the iterative embedding results. On the other hand, the results of the finite-field approach heavily depend on the density functional. When the best performing functionals are used, the finite-field approach yields isotropic and anisotropic polarizabilities in very good agreement with CCSD.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9376–9387"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00767","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
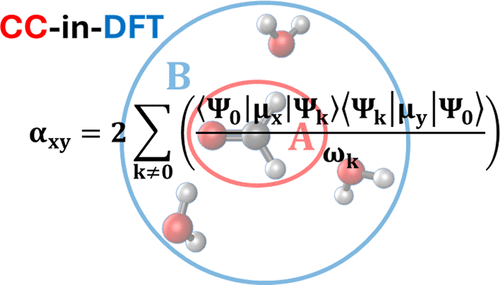
Abstract
We present a study of static polarizabilities of organic molecules in aqueous environments using projection-based coupled-cluster in density functional theory quantum embedding. We propose two methods for the computation of supermolecular polarizabilities: an iterative embedding approach and a finite-field approach. The performance of these methods is tested against regular coupled-cluster singles and doubles (CCSD) theory. The static polarizability tensor of the investigated organic molecules varies only slightly with the inclusion of water molecules. The iterative CCSD-in-DFT approach produces isotropic polarizabilities of CCSD quality with mean relative errors smaller than 1.0% at a reduced computational cost, while the anisotropic polarizabilities are not as well described. The choice of the exchange correlation functional for the treatment of the environment has little impact on the quality of the iterative embedding results. On the other hand, the results of the finite-field approach heavily depend on the density functional. When the best performing functionals are used, the finite-field approach yields isotropic and anisotropic polarizabilities in very good agreement with CCSD.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


