Samuel Murail, , , Jaysen Sawmynaden, , , Akli Zemirli, , , Maud Jusot, , , Fabio Pietrucci, , , Jacques Chomilier, , , Pierre Tufféry, , and , Dirk Stratmann*,
{"title":"Robust Conformational Space Exploration of Cyclic Peptides by Combining Different MD Protocols and Force Fields","authors":"Samuel Murail, , , Jaysen Sawmynaden, , , Akli Zemirli, , , Maud Jusot, , , Fabio Pietrucci, , , Jacques Chomilier, , , Pierre Tufféry, , and , Dirk Stratmann*, ","doi":"10.1021/acs.jctc.5c01123","DOIUrl":null,"url":null,"abstract":"<p >Cyclic peptides are an important class of pharmaceutical drugs. We used replica-exchange molecular dynamics (REMD) and simulated tempering (ST) simulations to explore the conformational landscape of a set of nine cyclic peptides. The N-ter to C-ter backbone-cyclized peptides of 7-10 residues were previously designed for high conformational stability with a mixture of <span>l</span>- and <span>d</span>-amino acids. Their experimental NMR structures are available in the protein data bank (PDB). For each peptide, we tested several force fields, namely, Amber96, Amber14, RSFF2C, and Charmm36m in implicit and explicit solvents. We find that the variability of the free energy maps obtained from several protocols is larger than the variability obtained by just repeating the same protocol. Running multiple protocols is therefore important for the convergence assessment of REMD or ST simulations. The majority of the free energy maps showed clusters with a high RMSD compared to the native structures, revealing the residual flexibility of this set of cyclic peptides. The high RMSD clusters had in some cases the lowest free energy, rendering the prediction of the native structure more difficult with a single protocol. Fortunately, the combination of four implicit solvent REMD and ST simulations, mixing the Amber96 and Amber14 force fields, predicted robustly the native structure. As implicit solvent simulations in the REMD or ST setup are up to one hundred times faster than explicit solvent simulations, running four implicit solvent simulations is a good practical choice. We checked that the use of an explicit solvent REMD or ST simulation, taken alone or combined with implicit solvent simulations, did not significantly improve our results. It results in our combination of four implicit solvent simulations being tied in terms of success rate with much more expensive combinations that include explicit solvent simulations. This may be used as a guideline for further studies of cyclic peptide conformations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"10018–10034"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01123","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
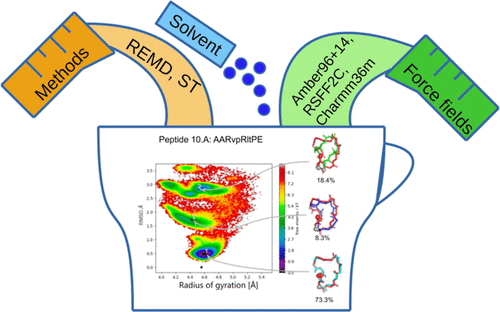
Cyclic peptides are an important class of pharmaceutical drugs. We used replica-exchange molecular dynamics (REMD) and simulated tempering (ST) simulations to explore the conformational landscape of a set of nine cyclic peptides. The N-ter to C-ter backbone-cyclized peptides of 7-10 residues were previously designed for high conformational stability with a mixture of l- and d-amino acids. Their experimental NMR structures are available in the protein data bank (PDB). For each peptide, we tested several force fields, namely, Amber96, Amber14, RSFF2C, and Charmm36m in implicit and explicit solvents. We find that the variability of the free energy maps obtained from several protocols is larger than the variability obtained by just repeating the same protocol. Running multiple protocols is therefore important for the convergence assessment of REMD or ST simulations. The majority of the free energy maps showed clusters with a high RMSD compared to the native structures, revealing the residual flexibility of this set of cyclic peptides. The high RMSD clusters had in some cases the lowest free energy, rendering the prediction of the native structure more difficult with a single protocol. Fortunately, the combination of four implicit solvent REMD and ST simulations, mixing the Amber96 and Amber14 force fields, predicted robustly the native structure. As implicit solvent simulations in the REMD or ST setup are up to one hundred times faster than explicit solvent simulations, running four implicit solvent simulations is a good practical choice. We checked that the use of an explicit solvent REMD or ST simulation, taken alone or combined with implicit solvent simulations, did not significantly improve our results. It results in our combination of four implicit solvent simulations being tied in terms of success rate with much more expensive combinations that include explicit solvent simulations. This may be used as a guideline for further studies of cyclic peptide conformations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


