Engineering unnatural amino acids in peptide linkers enables cathepsin-selective antibody-drug conjugates for HER2-positive breast cancer
IF 11.5
1区 医学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
Antibody-drug conjugates (ADCs) are a rapidly evolving class of targeted cancer therapeutics that combine the specificity of monoclonal antibodies with the potent cytotoxicity of small-molecule payloads. Their clinical success has led to significant advances in oncology, positioning ADCs as a transformative modality in cancer treatment. Most clinically approved ADCs utilize a protease-cleavable valine-citrulline (Val-Cit) dipeptide linker designed to facilitate intracellular payload release upon proteolytic activation by the lysosomal cathepsins. However, the Val-Cit linker is susceptible to off-target cleavage by proteases expressed in non-malignant tissues, resulting in premature payload release and systemic toxicity. To address this limitation, we established a high-throughput peptide linker discovery platform using Hybrid Combinatorial Substrate Library (HyCoSuL) screening to comprehensively profile protease substrate preferences. By incorporating unnatural amino acids, we identified peptide sequences with high selectivity toward cancer-associated proteases, thereby overcoming the constraints of conventional linker design. As proof of concept, we engineered trastuzumab-based ADCs selectively activated by cathepsin B or cathepsin L and evaluated their cytotoxic efficacy in HER2-positive breast cancer models. Our HyCoSuL-guided linkers display higher selectivity toward cathepsins than Val-Cit and enable faster, protease-dependent activation of peptide prodrugs and ADCs in vitro, while also exhibiting enhanced stability in human plasma to minimize premature payload release. Furthermore, recognizing that efficient protease-activated ADC function requires co-expression of both the target antigen and the activating protease, we performed single-cell mass cytometry analysis of patient-derived breast cancer samples to assess the correlation between HER2 and cathepsin expression. This analysis revealed heterogeneous cathepsin expression, underscoring that pairing the antibody target with the appropriate protease-selective linker might critical for robust, tumor-confined payload release. Our findings highlight the importance of patient stratification based on antigen and protease expression profiles, providing a foundation for developing ADCs with greater protease selectivity and minimized off-target activation.
Significance
This work presents a rational chemistry-driven strategy using unnatural amino acids to engineer peptide linkers with high cysteine cathepsins specificity and enhanced stability in human plasma. By combining comprehensive enzymatic profiling with single-cell CyTOF analysis of patient tumors, we outline a framework for precision-guided ADC design and protease-informed patient stratification in HER2-positive breast cancer.
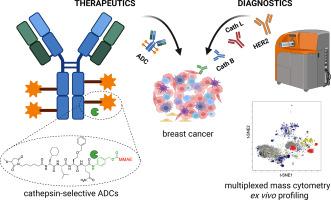
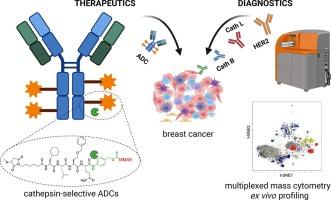
在肽连接体中设计非天然氨基酸,使组织蛋白酶选择性抗体-药物偶联物能够用于her2阳性乳腺癌
抗体-药物偶联物(adc)是一种快速发展的靶向癌症治疗药物,它结合了单克隆抗体的特异性和小分子有效载荷的强大细胞毒性。它们的临床成功导致了肿瘤学的重大进展,将adc定位为癌症治疗的变革性模式。大多数临床批准的adc使用蛋白酶可切割缬氨酸-瓜氨酸(Val-Cit)二肽连接物,用于在溶酶体组织蛋白酶的蛋白水解激活下促进细胞内有效载荷的释放。然而,Val-Cit连接体容易被非恶性组织中表达的蛋白酶脱靶切割,导致过早的有效载荷释放和全身毒性。为了解决这一限制,我们建立了一个高通量的肽连接物发现平台,使用杂交组合底物库(HyCoSuL)筛选来全面分析蛋白酶底物偏好。通过加入非天然氨基酸,我们确定了对癌症相关蛋白酶具有高选择性的肽序列,从而克服了传统连接体设计的限制。作为概念验证,我们设计了基于曲妥珠单抗的adc,通过组织蛋白酶B或组织蛋白酶L选择性激活,并评估了它们在her2阳性乳腺癌模型中的细胞毒性效果。与Val-Cit相比,我们的hycosol引导的连接物对组织蛋白酶具有更高的选择性,能够更快地在体外对肽前药和adc进行蛋白酶依赖性激活,同时在人血浆中也表现出更高的稳定性,以最大限度地减少过早的有效载荷释放。此外,认识到有效的蛋白酶激活ADC功能需要靶抗原和活化蛋白酶的共同表达,我们对患者来源的乳腺癌样本进行了单细胞细胞计数分析,以评估HER2和组织蛋白酶表达之间的相关性。该分析揭示了组织蛋白酶的异质表达,强调将抗体靶点与适当的蛋白酶选择性连接体配对可能对稳定的、肿瘤受限的有效载荷释放至关重要。我们的研究结果强调了基于抗原和蛋白酶表达谱的患者分层的重要性,为开发具有更高蛋白酶选择性和最小化脱靶激活的adc提供了基础。这项工作提出了一种合理的化学驱动策略,使用非天然氨基酸来设计具有高半胱氨酸组织蛋白酶特异性和增强血浆稳定性的肽连接物。通过将综合酶谱分析与患者肿瘤的单细胞细胞of分析相结合,我们概述了her2阳性乳腺癌中精确指导ADC设计和蛋白酶信息患者分层的框架。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Controlled Release
医学-化学综合
CiteScore
18.50
自引率
5.60%
发文量
700
审稿时长
39 days
期刊介绍:
The Journal of Controlled Release (JCR) proudly serves as the Official Journal of the Controlled Release Society and the Japan Society of Drug Delivery System.
Dedicated to the broad field of delivery science and technology, JCR publishes high-quality research articles covering drug delivery systems and all facets of formulations. This includes the physicochemical and biological properties of drugs, design and characterization of dosage forms, release mechanisms, in vivo testing, and formulation research and development across pharmaceutical, diagnostic, agricultural, environmental, cosmetic, and food industries.
Priority is given to manuscripts that contribute to the fundamental understanding of principles or demonstrate the advantages of novel technologies in terms of safety and efficacy over current clinical standards. JCR strives to be a leading platform for advancements in delivery science and technology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


