Exploring the guest–host relationship for zeolite Y: a synergistic structural and theoretical investigation
IF 9.5
2区 材料科学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Understanding the mobility and distribution of cations in the presence of additional guest species within the zeolite framework is inherently very complex because several thermal and chemical processes occur simultaneously. In this study, the dehydration of zeolite Na-Y was studied using in situ powder X-ray diffraction (XRD). The crystal structure parameters, both the evolution of lattice parameters and the occupancies of sodium at specific positions in the model, were monitored upon heating. A complementary computational study was performed to understand interactions between the framework, cations, and water at the molecular level. Grand Canonical Monte Carlo (GCMC) simulations provided initial insights into water adsorption. Machine Learning Potentials (MLPs) were then trained to the ab initio Potential Energy Surface (PES) using a deep neural network, modeling the dynamics of the framework and cations at various water loadings. Strong agreement between computational results and experimental data reveal that upon dehydration, zeolite Na-Y initially contracts due to water removal, but subsequently expands as sodium cations migrate to the double 6-membered rings (site I). This study demonstrates significant benefits of integrating parametric Rietveld refinement and Machine Learning assisted Molecular Dynamics simulations in understanding dynamic behavior of guest molecules in nanoporous materials at operating conditions and interpreting complex and convoluted experimental data.
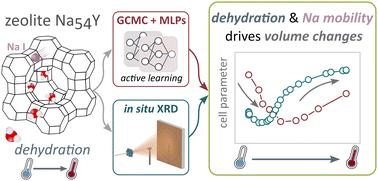
探索Y沸石的主客关系:一个协同结构和理论研究
在沸石骨架中存在额外客体时,了解阳离子的迁移率和分布本质上是非常复杂的,因为几个热和化学过程同时发生。本文采用原位粉末x射线衍射(XRD)对Na-Y沸石的脱水过程进行了研究。在加热时监测晶体结构参数,包括晶格参数的演变和钠在模型中特定位置的占有率。在分子水平上进行了一项互补的计算研究,以了解框架、阳离子和水之间的相互作用。大规范蒙特卡罗(GCMC)模拟提供了对水吸附的初步见解。然后使用深度神经网络将机器学习电位(mlp)训练为从头算势能面(PES),模拟各种水负荷下框架和阳离子的动态。计算结果和实验数据之间的强烈一致性表明,在脱水时,沸石Na-Y最初由于水的去除而收缩,但随后随着钠离子迁移到双6元环(位点I)而膨胀。该研究表明,在理解纳米多孔材料中客体分子在操作条件下的动态行为和解释复杂而复杂的实验数据方面,将参数Rietveld细化和机器学习相结合的分子动力学模拟具有显著的优势。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Materials Chemistry A
CHEMISTRY, PHYSICAL-ENERGY & FUELS
CiteScore
19.50
自引率
5.00%
发文量
1892
审稿时长
1.5 months
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


