Design, synthesis, anticancer evaluation, biological screening, and computational study of novel 6,7-dimethoxyquinazoline derivatives as VEGFR-2 inhibitors and apoptotic inducers
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
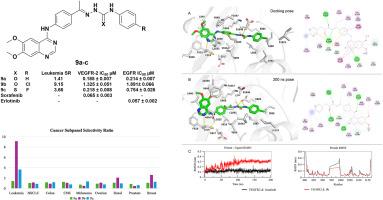
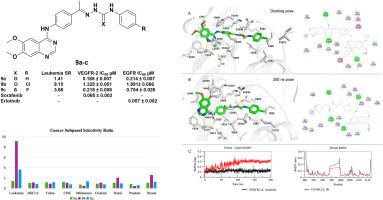
Novel quinazoline analogs were designed, synthesized, and evaluated for their activities as potential anticancer agents. The compounds bearing a 4-substituted-6,7-dimethoxyquinazoline scaffold underwent in vitro cytotoxicity screening against 60 cancer cell lines under the National Cancer Institute (NCI) protocol, for all the novel synthesized compounds at 10 μM concentration. Notably, the initial single-concentration screening revealed that compounds 9a-d exhibited the most potent growth inhibition percentage (GI%) on most of the screened nine panels of cancer, with a mean range of (50.23–189.98 %). The most potent compounds, 9a-c, continued to be tested at concentrations of 0.01, 0.1, 1, 10, and 100 μM to identify the GI50, TGI, and LC50 values and evaluate the compounds' efficacy and selectivity. Compound 9a generally demonstrated broad-spectrum potent growth inhibitory activity (GI50) reaching to nanomolar level (GI50 = 10 nM) against leukemia CCRF-CEM and colon COLO-205 cells. Compound 9b exhibited a leukemia subpanel midpoint inhibitory value (MIDb) of 0.56 μM, with a selectivity ratio (SR) of 9.15 against leukemia cell lines. Additionally, 9b induced apoptosis and caused cell cycle arrest at the G1 phase in leukemia CCRF-CEM cells. The RT-PCR results showed that 9b increased the levels of proapoptotic mediators, Bax, caspase-3, and p53, with values of 8.91, 6.15, and 4.95 fold change, respectively, in addition to the decreased level of anti-apoptotic protein Bcl-2 to 0.372 fold change in the treated CCRF-CEM leukemia cells. Compound 9b was docked to VEGFR-2 (ΔG = −14.1 kcal/mol), suggesting strong interactions within the VEGFR2 active site, comparable to the reference ligand sorafenib (ΔG = −14.8 kcal/mol). The molecular dynamic (MD) simulation (200ns) confirmed the docking results by demonstrating that compound 9b retained stable interactions inside the VEGFR-2 active site throughout the trajectory. Moreover, the MTT viability test for compounds 9a, 9b, and 9c demonstrated less cytotoxicity against normal fibroblast cells (WI38), revealing enhanced safety profiles with IC50 values of 28.04, 219.79, and 43.77 μM, respectively, compared to Sorafenib (IC50 = 26 μM). Enzyme inhibition assays revealed that compounds 9a-c effectively inhibited EGFR and VEGFR-2, confirming the multi-targeting potential of this series of compounds.
新型6,7-二甲氧基喹唑啉衍生物作为VEGFR-2抑制剂和凋亡诱导剂的设计、合成、抗癌评价、生物学筛选和计算研究
设计、合成了新型喹唑啉类似物,并对其潜在的抗癌活性进行了评价。采用4-取代-6,7-二甲氧基喹唑啉支架进行体外细胞毒性筛选,所有新合成的化合物在10 μM浓度下对60种癌细胞进行体外细胞毒性筛选。值得注意的是,最初的单浓度筛选显示,化合物9a-d对筛选的9组癌症中的大多数表现出最有效的生长抑制百分比(GI%),平均范围为(50.23-189.98%)。在0.01、0.1、1、10和100 μM的浓度下,继续测定最有效的化合物9a-c的GI50、TGI和LC50值,并评价化合物的药效和选择性。化合物9a对白血病CCRF-CEM和结肠癌COLO-205细胞普遍表现出广谱强效生长抑制活性(GI50=10nM),达到纳摩尔水平。化合物9b对白血病亚组中点抑制值(MIDb)为0.56 μM,对白血病细胞的选择性比(SR)为9.15。此外,9b在白血病CCRF-CEM细胞中诱导细胞凋亡并导致细胞周期阻滞在G1期。RT-PCR结果显示,9b使CCRF-CEM白血病细胞中促凋亡介质Bax、caspase-3和p53水平分别升高了8.91、6.15和4.95倍,抗凋亡蛋白Bcl-2水平降低了0.372倍。化合物9b与VEGFR-2对接(ΔG= -14.1 kcal/mol),表明在VEGFR2活性位点内存在强相互作用,与参考配体索拉非尼(ΔG= -14.8 kcal/mol)相当。分子动力学(MD)模拟(200ns)证实了对接结果,表明化合物9b在整个轨迹中在VEGFR-2活性位点内保持稳定的相互作用。此外,化合物9a、9b和9c对正常成纤维细胞(WI38)的细胞毒性较低,IC50值分别为28.04、219.79和43.77 μM,与Sorafenib (IC50 = 26 μM)相比,安全性增强。酶抑制实验显示,化合物9a-c有效抑制EGFR和VEGFR-2,证实了该系列化合物的多靶点潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


