Alexis Ralli*, , , Tim Weaving, , , Peter V. Coveney, , and , Peter J. Love,
{"title":"Bridging Quantum Chemistry and MaxCut: Classical Performance Guarantees and Quantum Algorithms for the Hartree–Fock Method","authors":"Alexis Ralli*, , , Tim Weaving, , , Peter V. Coveney, , and , Peter J. Love, ","doi":"10.1021/acs.jctc.5c00948","DOIUrl":null,"url":null,"abstract":"<p >In quantum chemistry, self-consistent field (SCF) algorithms define a nonlinear optimization problem, with both continuous and discrete components. In this work, we derive Hartree–Fock-inspired SCF algorithms that can be exactly written as a sequence of Quadratic Unconstrained Spin/Binary Optimization problems (QUSO/QUBO). We reformulate the optimization problem as a series of MaxCut graph problems, which can be efficiently solved using semidefinite programming techniques. This procedure provides performance guarantees at each SCF step, irrespective of the complexity of the optimization landscape. We numerically demonstrate the QUBO-SCF and MaxCut-SCF methods by studying the hydroxide anion OH<sup>–</sup> and molecular Nitrogen N<sub>2</sub>. The largest problem addressed in this study involves a system comprised of 220 qubits (equivalently, spin–orbitals). Our results show that QUBO-SCF and MaxCut-SCF suffer much less from internal instabilities compared with conventional SCF calculations. Additionally, we show that the new SCF algorithms can enhance single-reference methods, such as configuration interaction. Finally, we explore how quantum algorithms for optimization can be applied to the QUSO problems arising from the Hartree–Fock method. Four distinct hybrid-quantum classical approaches are introduced: GAS-SCF, QAOA-SCF, QA-SCF and DQI-SCF.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9511–9524"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00948","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
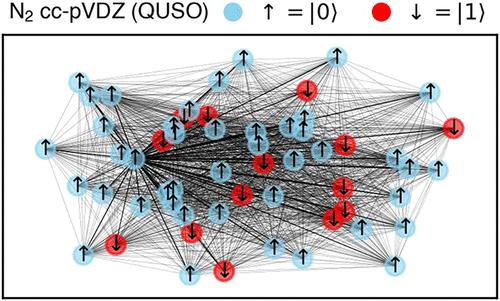
In quantum chemistry, self-consistent field (SCF) algorithms define a nonlinear optimization problem, with both continuous and discrete components. In this work, we derive Hartree–Fock-inspired SCF algorithms that can be exactly written as a sequence of Quadratic Unconstrained Spin/Binary Optimization problems (QUSO/QUBO). We reformulate the optimization problem as a series of MaxCut graph problems, which can be efficiently solved using semidefinite programming techniques. This procedure provides performance guarantees at each SCF step, irrespective of the complexity of the optimization landscape. We numerically demonstrate the QUBO-SCF and MaxCut-SCF methods by studying the hydroxide anion OH– and molecular Nitrogen N2. The largest problem addressed in this study involves a system comprised of 220 qubits (equivalently, spin–orbitals). Our results show that QUBO-SCF and MaxCut-SCF suffer much less from internal instabilities compared with conventional SCF calculations. Additionally, we show that the new SCF algorithms can enhance single-reference methods, such as configuration interaction. Finally, we explore how quantum algorithms for optimization can be applied to the QUSO problems arising from the Hartree–Fock method. Four distinct hybrid-quantum classical approaches are introduced: GAS-SCF, QAOA-SCF, QA-SCF and DQI-SCF.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


